












【佳学基因检测】多发性骨髓瘤的深度基因检测及大数据分析
血液肿瘤基因检测导读:
为了研究 lrWGS 是否可用于识别多发性骨髓瘤中的反复和私人遗传畸变,多发性骨髓瘤的深度基因检测及大数据分析对一组 37 个多发性骨髓瘤样本进行了 lrWGS。采集所有骨髓样本用于诊断目的(诊断时 37 例中的 35 例),并采用扩展的 FISH 检测包进行分析。为确保对纯的细胞亚群进行分析,所有使用的细胞都进行了 FACS(图 1A). 此外,通过主要对 T 细胞执行 lrWGS,在 37 个病例中的 32 个病例中生成了种系对照。准备好的 lrWGS 文库在多发性骨髓瘤样本中测序深度为 31.2× 和 胚系对照样本测序深度平均为31.6× 的平均深度。
 样本中接受 FACS 的变性细胞上进行.jpg)
图1:lrWGS 可以直接在从诊断骨髓 (BM) 样本中接受 FACS 的变性细胞上进行。(A) 实验过程的示意图概述。为了进行遗传、转录和表观遗传学表征,对来自患者骨髓样本的多发性骨髓瘤和正常细胞(T 细胞 [T] 或粒系细胞 [My])进行 FACS。lrWGS 文库直接在变性细胞上制备,无需事先进行 DNA 纯化。(BD) 散点图 (B) 和箱线图 (CD) 显示了使用指定的细胞类型和方法制备的 lrWGS 文库的 N50 相块大小和平均 DNA 分子长度。显示了从 10X 平台上制备的 DNA(HMW 或柱纯化)生成的已发布 lrWGS 的质量控制数据以供比较。
可以从少量变性细胞制备高质量的 lrWGS 文库
要充分利用 lrWGS 技术,必须从纯化的 HMW DNA 中生成文库。为了避免 HMW 制备的需要,佳学基因开发了一个实验方案,用于直接在用氢氧化钠变性的细胞上制备 lrWGS 文库。这使佳学基因能够在 Chromium Genome 平台上对 200 至 240 个接受 FACS(包含 ~1.2 ng DNA)的细胞制备 lrWGS 文库。佳学基因发现,这个简单的实验方案允许维持非常大的 DNA 分子(中位 DNA 片段长度,216 kbp),这直接反映在重建相块的大小(中位 N50,14.8 Mbp;中位最长相块,60.2 Mbp ;图 1B-D)。最长的组装相位块为 120.9 Mbp,几乎相当于 4 号染色体的整个 q 臂(138.4 Mbp 中的 120.9)。因此,该相块几乎达到了在不穿过着丝粒区域的情况下可实现的理论最大长度。正如预期的那样,多发性骨髓瘤样本通常在杂合性缺失的区域显示出较差的定相,但总体而言,多发性骨髓瘤 样本的 N50 相位块大小与正常对照相当(图 1B-C)。 将佳学基因的 lrWGS 数据与从 10X 平台上纯化的 HMW DNA 生成的高质量数据集进行比较,佳学基因的文库是从明显更长的 DNA 分子生成的,并保持相似的相块大小(图 1B-D).
总的来说,多发性骨髓瘤的深度基因检测及大数据分析得出结论,直接在变性细胞上进行 lrWGS 文库制备可保持 DNA 完整性,并允许从数量非常有限的接受 FACS 的细胞中生成高质量的 lrWGS。
lrWGS 和 FISH 鉴定的 CNV 高度一致
CNV 是多发性骨髓瘤的一个共同特征,用来区分HRD 亚组和高危患者。为识别 CNV,我们计算了拷贝数,生成了CNV数据。比较从 lrWGS 数据计算的染色体数据与从 FISH 计算的染色体数据,多发性骨髓瘤的深度基因检测及大数据分析发现在 37 个多发性骨髓瘤中的 35 个(95%)中正确预测了染色体数目。未正确计算拷贝数的 2 个案例显示了广泛而复杂的亚克隆拷贝数变化。没有 CNV 调用程序(包括 FindSV、CNVkit、 ASCAT、和 Battenberg 方法) 正确地预测了这 2 个样本中的染色体数。尽管如此,根据 4 号染色体覆盖率(FISH 分析中唯一已知具有正常拷贝数的染色体)校正拷贝数,可以目视识别 FISH 发现的所有(12 条中的 12 条)CNV(图 2A). 在 lrWGS 正确预测染色体数目的 35 名患者中,FISH 鉴定的所有 CNV 的 96%(149 名中的 143 名),包括所有 1q 扩增,在 lrWGS 数据中得到了一致性的结果(图 2A-B). 这些结果与以前旨在比较 FISH 与传统 WGS 以鉴定 CNV 的研究结果相似。

图 2:lrWGS 允许高效地检测 CNV 和识别双重打击多发性骨髓瘤。(A) 热图显示根据 lrWGS 覆盖范围计算的体细胞染色体 (chr1-22) 的拷贝数。拷贝数在两种情况下(由 * 表示)更正为 chr4 拷贝数(补充方法)。垂直黑线表示 FISH 探针的基因组定位。FISH 研究的区域用黑点表示,点在灰线上方或下方的位置分别表示拷贝数增加或减少。(B) 维恩图显示 FISH 调用的 CNV 和 FISH 面板中包含的区域(不包括 IGH 区域)中的 lrWGS 调用的 CNV 之间的重叠。FISH 调查的区域显示在面板 A 中。(C) TP53 基因座周围区域中总的、单倍型特定的和未定相的 lrWGS 覆盖范围的拷贝数。显示正常对照样本(种系 [GL])中的中值拷贝数以供比较。垂直线表示用于识别 chr17p CNV 的 FISH 探针的边界。对于 P11603_109,指示了获得的 TP53 突变(使用 lrWGS 数据检测)的位置。VAF,变异等位基因频率。
总的来说,这表明 lrWGS 可以高效地识别 CNV。然而,多发性骨髓瘤的深度基因检测及大数据分析的数据表明,应谨慎验证 WGS 在具有潜在广泛和复杂的亚克隆拷贝数变化的肿瘤中预测的染色体数。
识别双重打击TP53 失活
多发性骨髓瘤中的双重打击TP53 失活与不良结果相关,即使采用新疗法也是如此。这促使佳学基因仔细调查多发性骨髓瘤的深度基因检测及大数据分析的患者队列中是否存在此类事件。在分阶段 lrWGS 数据中可以很容易地观察到单倍型特异性染色体 17p 缺失(图 2C)。基于 CNV 的计算调用,多发性骨髓瘤的深度基因检测及大数据分析确定了 8 名 CNV 患者中的 7 名 (87.5%) 涉及TP53基因座。通过 FISH 在 24% 的细胞中发现了 lrWGS 数据中鉴定的克隆参与最低的 17p 缺失。相比之下,仅在 14% 的细胞中发现了 FISH 鉴定的未检测到的 17p 缺失。接下来,多发性骨髓瘤的深度基因检测及大数据分析使用 Sarek 确定了获得性突变,并分析了多发性骨髓瘤中反复突变的基因的序列变化。多发性骨髓瘤的深度基因检测及大数据分析确定了 2 名移码突变对 TP53 功能有害的患者。第一个是亚克隆的,出现在 TP53 位点 (P13756_103) 正常的患者身上。相比之下,第二个突变接近克隆(存在于患者 P11603_109 ≥ 80% 的读数中)并与克隆 17p 缺失同时发生(图 2C, 右), 表明该患者在诊断时表现为高风险双重打击 TP53多发性骨髓瘤。
原发生发性骨髓瘤IGH 基因座易位很容易通过 lrWGS 识别
SV 可以在 lrWGS 数据中识别为跨越染色体断点周围区域的读取云的富集(图 3A)。这可用于识别易位、模板化插入、局部放大和其他复杂的 SV(图 3B-C)。多发性骨髓瘤的深度基因检测及大数据分析使用 Long Ranger 和 GROCS-SVs 来调用染色体间 SV。这种方法在所有样本中识别出 198 个 SV(图 4A)。尽管 3 例 HRD多发性骨髓瘤未检测到 SV,但在 HRD 和非 HRD 病例之间未发现每个样本的结构性变异(SV)数量存在显着差异(图 4B).
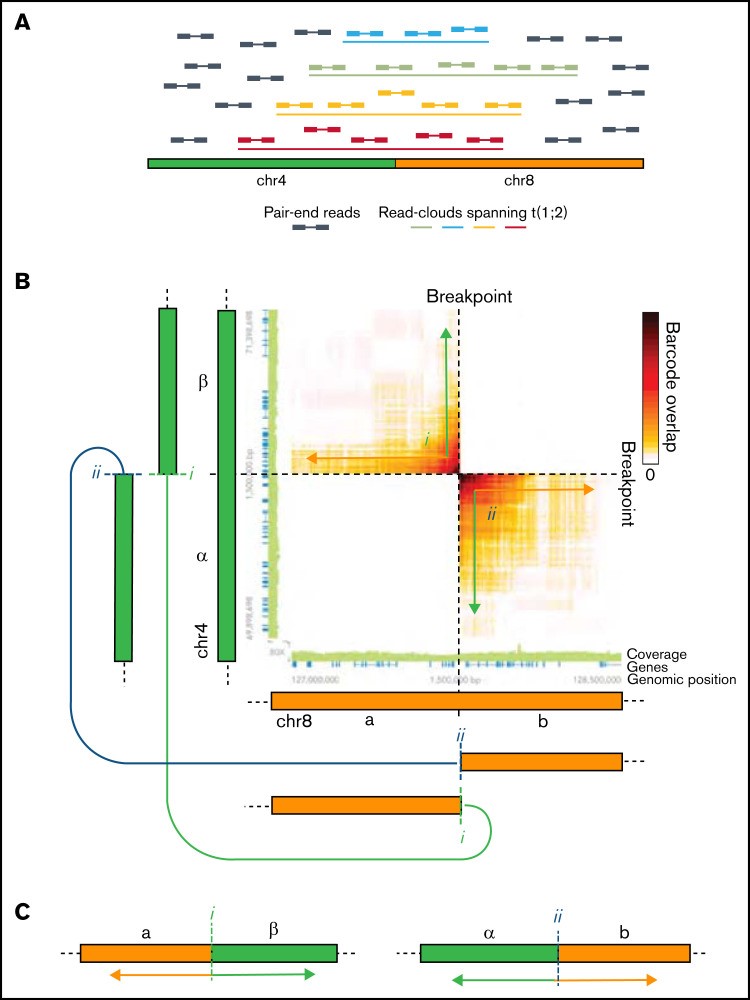
图 3:SV 在 lrWGS 中被识别为跨断点读取云的丰富。(A) 跨越 4 号染色体 (chr4) 到 chr8 易位断点的读取云和构成读取云的对端读取的示意图。(B) 热图显示通过相互易位融合的 chr4 和 chr8 区域之间共享读取云(条形码)的丰富。随着共享读取云的富集逐渐增加,接近断点,热图中的信号类似于指向断点的 2 个箭头。实际上,这允许确定相互易位导致 chr4 β-区域与 chr8 a-区域 (i) 以及 chr4 α-区域与 chr8 b-区域 (ii) 的融合。连接的绿色和橙色箭头示意性地表示事件 i 和 ii 连接的区域。(C) 相互易位产生的衍生染色体示意图。
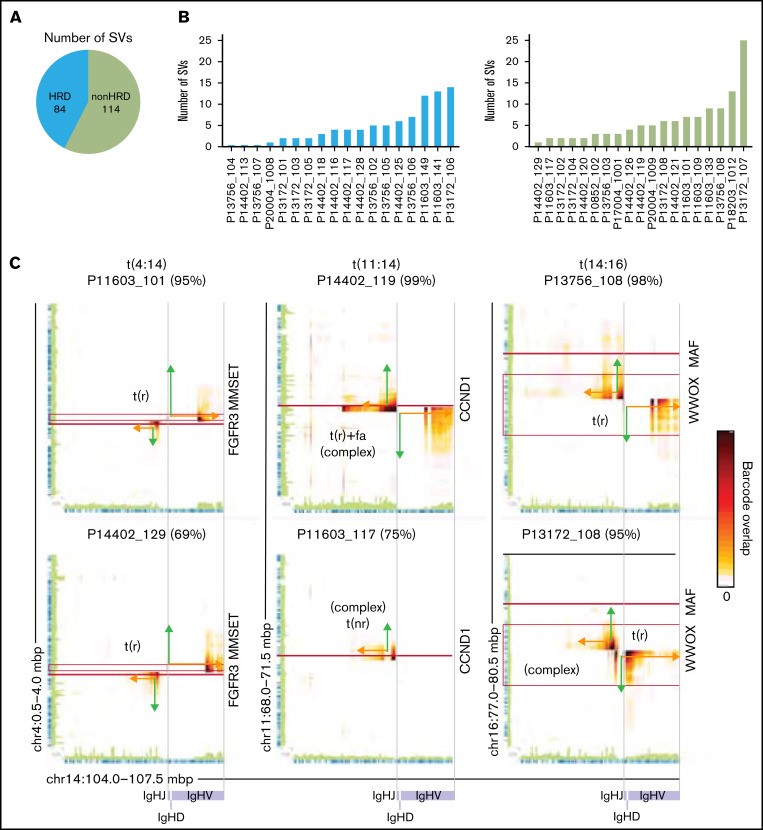
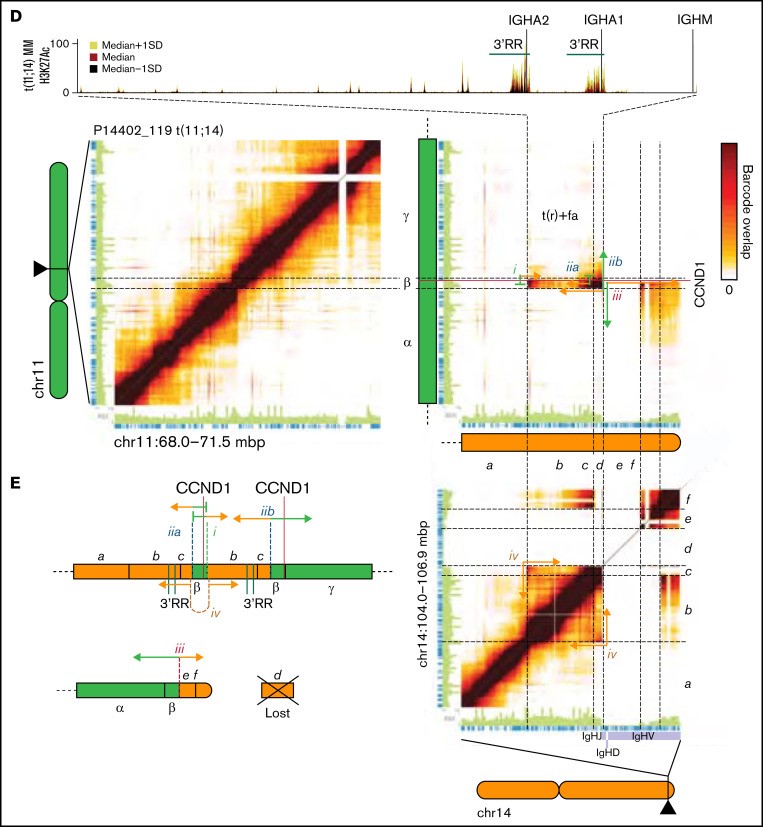
图 4:跨断点读取云允许识别和重建 IGH 易位事件。(A) 涉及 HRD 和非 HRD多发性骨髓瘤中鉴定的不同染色体的结构性变异(SV)总数。(B) HRD(左)和非 HRD 多发性骨髓瘤(右)中涉及不同染色体的结构性变异(SV)数量。(C) 热图显示 IGH 基因座(染色体 14 [chr14])和 chr4 (多发性骨髓瘤SET)、chr11 (CCND1) 和 chr16 (MAF) 上的断点区域之间共享的读取云数。连接的绿色和橙色箭头示意性地表示易位连接的区域。SV 的类型显示在读取云簇旁边。如果所描绘的结构性变异(SV)是更复杂结构性变异(SV)的一部分,则会指出这一点。FISH 发现的携带指定易位的细胞百分比显示在每个热图上方的括号中。水平(红色)和垂直(紫色灰色)线分别标记指定基因和 IGH VDJ 区域的位置。(D) P14402_119 中 chr11(左上)、从 chr11 到 chr14(右上)和 chr14(右下)的读取云重叠。轨道以 t(11;14)多发性骨髓瘤显示 H3K27Ac 信号(中值±标准偏差 [SD])。连接的绿色和橙色线示意性地表示事件 i 到 iv 连接的区域。(E) P14402_119 中衍生染色体的示意图,由 lrWGS 确定。t(nr),单向易位;t(r),相互易位; t(r)+fa,相互易位与断点区域的局灶性放大。
为了识别 IGH 易位,多发性骨髓瘤的深度基因检测及大数据分析使用Loupe可视化了常见的 t(4;14)、t(11;14) 和 t(14;16) 断点区域。多发性骨髓瘤的深度基因检测及大数据分析发现易位可以很容易地通过跨断点读取云的丰富在视觉上识别(图 4C; )。富集对单个 IGH 易位事件具有高度特异性,并且可以清楚地与背景信号区分开来。在计算上称为 SV中,多发性骨髓瘤的深度基因检测及大数据分析发现 16 个多发性骨髓瘤具有涉及多发性骨髓瘤SET、CCND1或MAF基因座的 IGH 易位。总体而言,在 FISH 识别的 17 个 IGH 易位中,有 16 个(94%)被 lrWGS 发现,并且没有出现假阳性。未识别的 t(11;14) 是亚克隆的(28% 的细胞),并且在目视检查时识别出支持结构性变异(SV)存在的读取云。
总之,这表明 lrWGS 允许正确识别 SV,包括常见的 IGH 易位。
t(11;14) 与 3'RR IGH 增强子区域的扩增有关
根据 IGH 易位事件经常由于错误的类别转换重组而发生的观点, IGH 易位位于 IGHJ 区域的着丝粒侧(图 4C)。具体观察 t(4;14) 和 t(14;16),多发性骨髓瘤的深度基因检测及大数据分析发现它们都是相互的,14 号染色体断点发生在 Eμ 和 3'RR 增强子区域(侧翼 IGHA1/2 区域). 同样,大约一半(9 个中的 5 个)t(11;14) 事件是常规易位(图 4C)将 3'RR 与 CCND1 基因座并列。有趣的是,1 个 t(14;16)相互易位和 3 个 t(11;14) 是复杂结构性变异(SV)的一部分,在 IGH 基因座外有额外的断点连接。
剩余的 t(11;14) 多发性骨髓瘤(9 个中的 4 个)呈现读取云簇模式,表明影响断点区域的更复杂事件(图 4C)。其中三名患者(P14402_119、P17004_1001 和 P13172_102)具有相似的模式,具有 3 个重叠的读取云簇。分析 P14402_119 中克隆 t(11;14) 的读数云重叠,多发性骨髓瘤的深度基因检测及大数据分析发现该模式与相互易位一致 (图 4Di-iib)在携带CCND1的衍生染色体上携带易位断点区域的焦点扩增(图 4Dii-iv,E). 这可以通过 11 号染色体上的拷贝数增加来推断(图 4D, β 区)和 14 号染色体(图 4D, bc 区域), 14 号染色体上扩增区域的着丝粒 (5') 和端粒 (3') 末端之间的共享读数云 (图 4Div, bc 区域),读取云在中间重叠与 2 个独立事件兼容,其中 1 个在 β 区域末端附近突然结束,重叠读取云没有逐渐减少(图4Diia) 和 1 显示了一个传统模式,随着离断点距离的增加,共享读取云的数量逐渐减少 (图 4Diib). 其他 2 个具有类似读取云模式且涉及重叠区域的案例(P17004_1001 和 P13172_102)可以用相同的方式解决。此外,P14402_119 中的 IGH 易位是复杂结构性变异(SV)的一部分,在 IGH 和 CCND1 基因座之外发生额外的易位(补充图 9)。观察第四个案例 (P14402_120),它有一个不同的模式,有 2 个(在基因组位置方面)部分重叠的读取云簇,多发性骨髓瘤的深度基因检测及大数据分析得出结论,这个模式代表了在焦点放大内发生的相互易位,导致重复的侧翼区域在易位断点的任一侧(补充图 10)。将涉及的区域与 H3K27Ac(标记活性基因调控元件)ChIPseq 数据重叠,图 4D)。正如预期的那样,这与CCND1的非常高的表达相关(所有 4 名患者均 > 1000 TPM)。
总之,这表明 t(11;14) 事件通常与CCND1基因座和 IGH 基因座的 3'RR 的扩增有关。此外,正如预期的那样,IGH 易位常常是复杂结构变 划的(17 个案例中的 5 个)的一部分。
lrWGS 允许检测和解析影响 MYC 基因座的各种 SV
MYC易位已被证明与不良结果有关。涉及MYC基因座的SV通常很复杂,涉及局部扩增、插入和易位,基因解码认为易位使得基因位于强增强子元件附近从而使MYC表达失调。依靠结构性变异(SV)的计算识别,多发性骨髓瘤的深度基因检测及大数据分析发现结构性变异(SV)影响37 名患者中的 9 名(24%)的MYC基因座,IGL 区域是唯一反复涉及的基因座(2 9 例)。已识别的结构性变异(SV)由不同的读取云集群支持(图 5A) 可以很容易地从背景中区分出来,在 1 个案例 (P14402_117) 中,该事件是更复杂的结构性变异(SV)的一部分。大多数已识别的MYC SV 是简单易位(9 个中有 5 个;图 5A). 仔细观察具有重叠读取云簇的其余案例,多发性骨髓瘤的深度基因检测及大数据分析得出结论,这些案例代表了带有模板插入的焦点放大(图 5A-B) 或相互易位 (图 5A、C). 这两种结构性变异(SV)在MYC基因座都进行了分析。正如预期的那样,基因解码发现的结构性变异(SV)影响包含MYC和PVT1启动子的区域(9 例中的 6 例)或PVT1基因的端粒区域(9 例中的 3 例),这此区域都含有影响MYC的元素基因。影响 PVT1 侧翼增强子区域的所有 3 个结构性变异(SV)通过插入(P14402_118 和 P11603_101;图 5B)或通过相互易位(P13756_108;图 5C)导致该元件与来自另一条染色体的骨髓瘤活性基因调控元件并置。同样,在大多数情况下,影响MYC / PVT1启动子区域的易位导致MYC基因座与来自不同基因组位置的骨髓瘤活性元件并列,包括 JCHAIN 和 IGL 基因座。总体而言,具有MYC SV 的多发性骨髓瘤表达的MYC水平高于具有正常MYC基因座的多发性骨髓瘤病例,但差异无统计学意义。
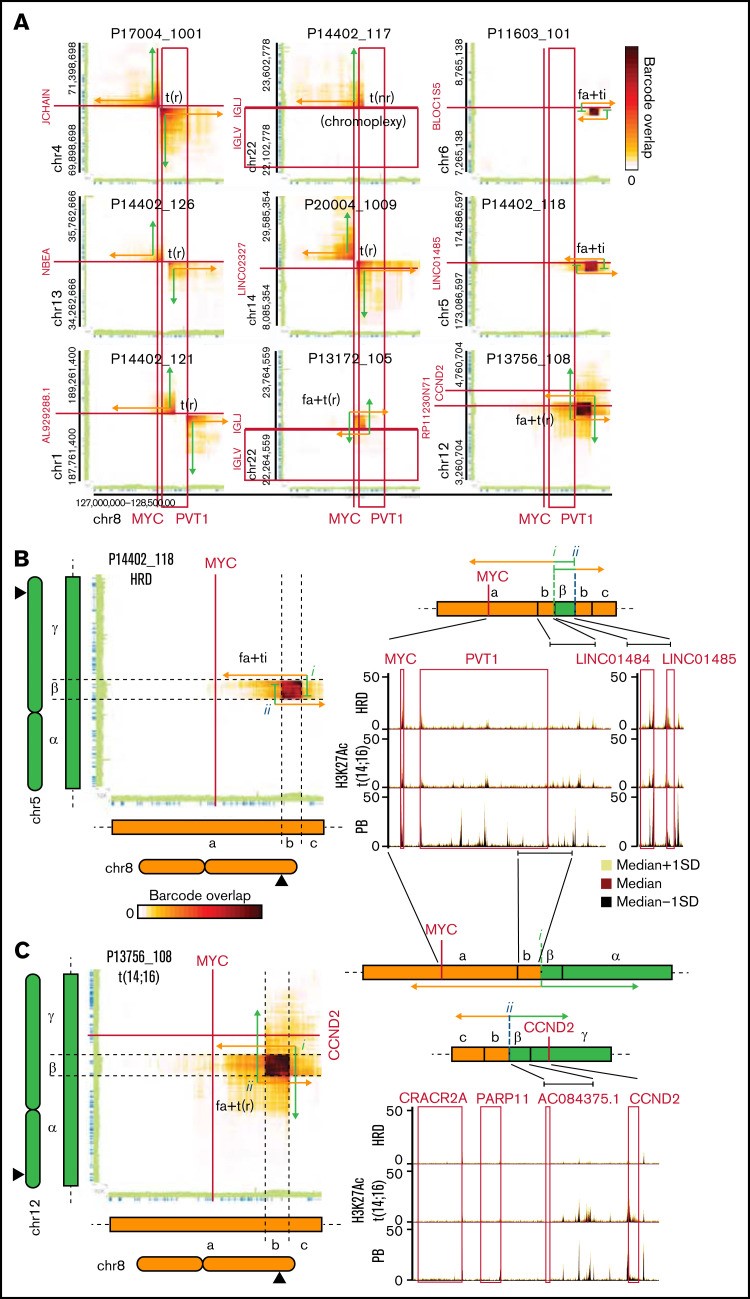
图 5:lrWGS 可以识别影响 MYC 基因座的各种 SV。(A) 热图显示 MYC 基因座和指示的基因组位置之间共享的读取云的数量。显示了来自检测到影响基因组 MYC 区域的结构性变异(SV)的患者 (n = 9) 的数据。连接的绿色和橙色箭头示意性地表示结构性变异(SV)连接的区域。SV 的类型显示在读取云簇旁边。图 3 提供了有关读取云模式解释的详细信息。如果所描绘的结构性变异(SV)是更复杂结构性变异(SV)的一部分,则会指出这一点。(BC) P14402_118 (B) 中 chr8 和 chr5 或 P13756_108 (C) 中 chr12 上的 MYC 基因座之间的读取云重叠。在面板 BC 中,指出了用于解析衍生基因座结构的特征 (i-ii);右侧显示了衍生染色体区域的示意图;和轨迹显示指定多发性骨髓瘤亚型和浆母细胞 (PB) 中涉及区域的 H3K27Ac 信号(中值±标准偏差 [SD])。fa+ti,在局灶性放大中发生的模板化插入;fa+t(r),局灶性放大中发生的相互易位;t(nr),单向易位;t(r),相互易位。
总之,这表明MYC基因座受到多发性骨髓瘤中各种结构性变异(SV)的影响,并且可以使用 lrWGS 检测和结构解析这些 SV。
个体特异性结构性变异(SV)导致 MAFA 和 MAP3K14 的过度表达
为了确定是否可以找到可能代表潜在主要或高风险变异的其他 SV,多发性骨髓瘤的深度基因检测及大数据分析调查了是否可以识别影响先前被发现是 IGH 基因座易位伙伴的位点的私有 SV。这确定了 P13172_104 中涉及MAFA基因座的 at(1;8) 和涉及MAP3K14基因座的 P13172_101 中的 at(6;17) 。
患者 P13172_104 有 2 个 SV,涉及 1 号和 8 号染色体,第二个并列MAFA和 TENT5C 位点(图 6A-C). 对断点区域中读取云的分析表明,涉及MAF的 SV是克隆的。仔细观察断点,多发性骨髓瘤的深度基因检测及大数据分析发现MAFA基因与通常位于TENT5C基因侧翼的强多发性骨髓瘤活性增强子区域并列(图 6C). H3K27Ac ChIPseq 证实TENT5C侧翼增强子和MAFA近端元件在 P13172_104 的多发性骨髓瘤细胞中都具有高度活性。因此,这强烈表明 TENT5C 增强子导致 MAFA 基因的激活。与此一致,多发性骨髓瘤的深度基因检测及大数据分析在 P13172_104 的多发性骨髓瘤细胞中发现了非常高的MAFA表达(~1000 TPM)(图 6D). 鉴于 MAFA 是 Maf 转录因子家族的一部分,多发性骨髓瘤的深度基因检测及大数据分析接下来想要评估MAFA过度表达是否会导致类似于 t(14;16)(MAF过度表达)多发性骨髓瘤 中的基因调控情况。查看多发性骨髓瘤亚组特定的 H3K27Ac 信号,所有样本的层次聚类显示 P13172_104 与 t(14;16)多发性骨髓瘤聚类并与MAF过表达组共享 H3K27Ac 特征(图 6E). 因此,在 P13172_104 中看到的私有 t(1;8) 易位导致 MAFA 的过表达和 MAF 型基因调控景观的建立。
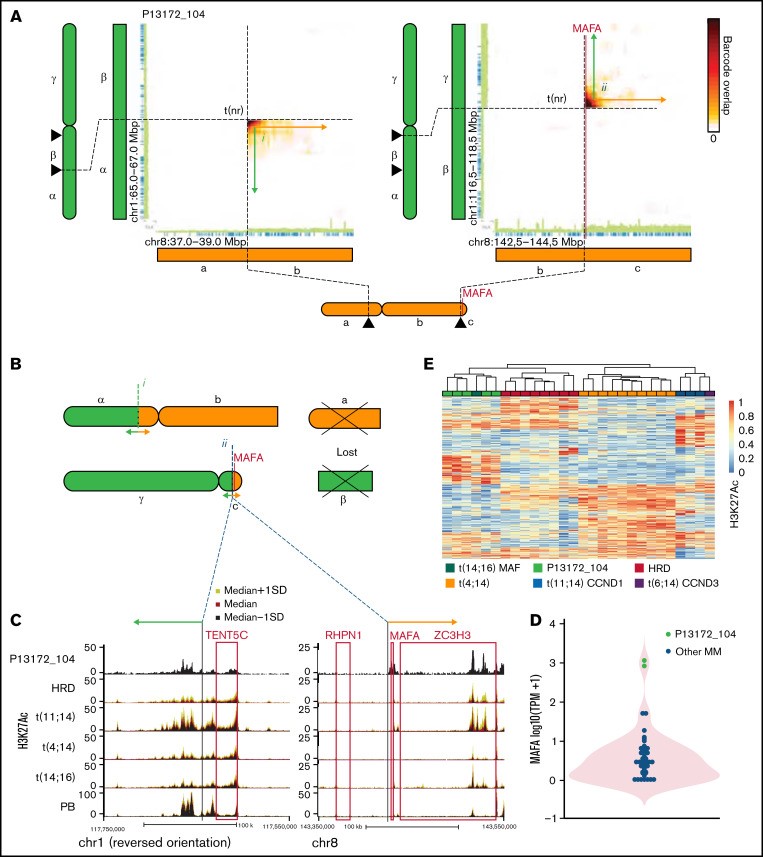
图 6:私有 SV 导致大量 MAFA 过表达并重新编程为 MAF 型多发性骨髓瘤。(A) 热图显示 1 号染色体 (chr1) 和 chr8 上指示区域之间共享的读取云数。连接的绿色和橙色箭头示意性地表示结构性变异(SV)连接的区域。指示结构性变异(SV)的类型。(B) 衍生染色体的示意图。(C) 在 t(1;8) 涉及的区域中显示 H3K27Ac 信号(中值±标准偏差 [SD])的轨道。(D) MAFA 的表达。(E) H3K27Ac 标记区域的行归一化层次聚类热图,显示多发性骨髓瘤亚型之间的差异信号。PB,浆母细胞;t(nr),单向易位。
影响MAP3K14基因座的结构性变异(SV)在结构上构成了 17 号染色体上的局灶性扩增,携带 6 号染色体小区域的模板化插入(图 7A-B). 读取云重叠分析表明结构性变异(SV)是亚克隆的。分析相关区域,多发性骨髓瘤的深度基因检测及大数据分析发现结构性变异(SV)将通常位于 BMP6 基因侧翼的增强子区域与 17 号染色体上的扩增区域并列(图 7B). 后者包含MAP3K14基因的一部分,但缺少最 3' 外显子,表明扩增不会产生该基因的 2 个功能拷贝。与失调 MAP3K14 基因的BMP6侧翼元件一致,P13172_101 中的表达很高(>300 TPM),几乎比任何其他研究的多发性骨髓瘤高 10 倍(图 7C). 由于MAP3K14作为NF - κB的激活剂发挥作用,这可能表明携带该结构性变异(SV)的多发性骨髓瘤亚克隆可能具有升高的或组成型的 NF-κB 信号传导。
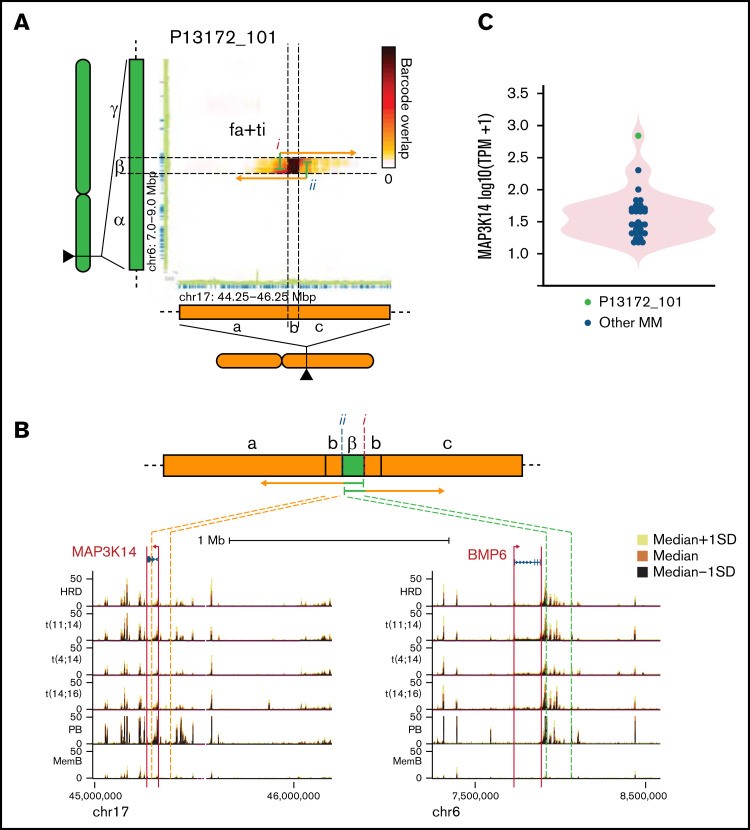
图 7:BMP6 侧翼增强子区域的插入导致 MAP3K14 的过度表达。(A) 热图显示 6 号染色体 (chr6) 和 chr17 上指示区域之间共享的读取云数。连接的绿色和橙色箭头示意性地表示结构性变异(SV)连接的区域。指示结构性变异(SV)的类型。(B) 派生染色体的示意图和显示结构性变异(SV)所涉及区域中 H3K27Ac 信号(中值±标准偏差 [SD])的轨道。(C) MAP3K14 的表达。fa+ti,发生在局灶性放大中的模板化插入。
多发性骨髓瘤的深度基因检测及大数据分析讨论
对于大多数患者而言,多发性骨髓瘤仍然是一种无法治好的疾病,预后不佳。因此,制定在临床常规中提供全面基因解码基因检测的策略对于了解个体患者的疾病和建立旨在改善结果和生存的个性化医疗计划具有至关重要的意义。
在这里,多发性骨髓瘤的深度基因检测及大数据分析进行了一项涉及 37 名多发性骨髓瘤患者的原理验证研究,并证明 lrWGS 可用于提供多发性骨髓瘤的综合基因解码基因检测信息。引人注目的是,lrWGS 通过简单地可视化跨越公共断点区域的读取云,提供了对经常性 IGH 和 IGL 易位的正确和直接的识别。鉴于读取云簇的特殊性,即使没有计算分析(除了基础处理)或种系控制,也可以快速识别这些 SV。在临床环境中,这很有吸引力,因为它允许在更耗时的整体分析之前快速筛选常见 SV。例如影响 IGH 和MYC的 SVloci,读取云簇的分析可以比 FISH 或传统 WGS 更简单、更正确地识别和解析不同的 SV。总的来说,考虑到 lrWGS 和 FISH 之间的高度一致性,这表明 lrWGS 可以作为一个独立的检测,可以减少用光学映射或其他旨在映射结构性变异(SV)的技术来补充基于测序的分析的需要。
利用对 lrWGS 数据中的遗传畸变进行整体分析的可能性,多发性骨髓瘤的深度基因检测及大数据分析确定了与不良结果相关的反复性和个体物异性事件,这些事件在当前的临床常规遗传学中不能被基于数据库比对的基因检测发现。在反复性变异中,多发性骨髓瘤的深度基因检测及大数据分析发现了 t(8;22) MYC-IGL 易位、高风险双重打击 TP53 失活和最近与不良结果相关的染色体碎裂的病例。有趣的是,多发性骨髓瘤的深度基因检测及大数据分析还可以发现导致基因过度表达的个体独特性结构变异,这些基因先前被证明是 IGH 基因座的易位伴侣(MAP3K14和MAFA)。特别是,涉及 MAFA 基因的 t(1;8) 是值得注意的,因为该结构变异可能代表一个替代的主要事件,其分子等同于具有相似患者风险特征和先天性的MAF的 t(14;16) 放松管制对蛋白酶体抑制剂的抗性。此外,多发性骨髓瘤的深度基因检测及大数据分析发现具有 t(11;14) 的样本通常携带 3'RR 和 CCND1 区域的重复。所分析的 t(11;14)多发性骨髓瘤是随机包含的事实表明这是一种相当普遍的现象,在复杂结构性变异(SV)插入期中有和没有衍生 t(11;14) 染色体参与的情况下都可以发现这种现象
名义上,lrWGS 可以在非常有限数量的细胞(≤1000 个细胞或 1-5 ng DNA)上进行,但对 HMW DNA 制剂的需求有效地阻止了这一点。正如多发性骨髓瘤的深度基因检测及大数据分析所做的那样,避免 DNA 制备的需要,以充分利用对输入材料的低要求,这对如何在多发性骨髓瘤和其他血液恶性肿瘤中进行常规基因解码基因检测具有有趣的意义。通过在 FACS 中运行诊断流式细胞术,可以对任何明确的肿瘤或正常细胞群进行基于 lrWGS 的基因解码分析。鉴于所需的细胞数量较少,这甚至可以在预期肿瘤细胞很少或数量较少的样本上进行,包括来自细针活检的样本或为最小残留疾病分析采集的样本。这种方法将同时允许对纯靶细胞群在进行深度基因检测分析的同时进行 RNA 测序和其他分析。最近引入的自动化程度更高的 FACS 有可能使这种分析工作流程在诊断流式细胞仪实验室中实施变得可行。
尽管从市场上移除 Chromium Genome 无疑阻碍了 lrWGS 的更广泛引入,但已经出现了其他几个平台来取而代之,包括单管长片段读取、转座酶联长读取测序、和液滴条形码测序。虽然必须进行更大规模的验证研究以证明这些可以有效地取代 10X 平台,但转座酶酶联长读长测序实验方案的简单工作流程使其成为潜在临床实施的有趣候选者。
总之,lrWGS 可以相对轻松地识别和解析不同结构性变异(SV)的结构,使其成为在多发性骨髓瘤和其他具有复杂基因组的血液恶性肿瘤中提供全面的基因检测分析的有吸引力的解决方案。此外,在诊断中使用 FACS 可以促进纯肿瘤细胞的 lrWGS 以及表达和表观遗传水平的基因测序分析。这可以为了解个体患者的潜在基因检测基础和疾病机制提供一个强大的平台。
- 【佳学基因检测】遗传性血管性水肿基因检测案例介绍...
- 【佳学基因检测】组织细胞增生症的基因检测及其基因突变根源的介绍...
- 【佳学基因检测】血液学癌症中RNA结合蛋白的小分子抑制药物...
- 【佳学基因检测】正确理解血友病基因检测...
- 【佳学基因检测】什么人要做Diamond-Blackfan贫血(先天再生障碍性贫血)基因解码、基因检测?...
- 【佳学基因检测】NK细胞形态异常基因检测...
- 【佳学基因检测】NK细胞自然(Nature)杀伤细胞缺乏基因检测...
- 【佳学基因检测】IgA肾病易感性3型基因解码、基因检测怎么预约解读?...
- 【佳学基因检测】多发性骨髓瘤的深度基因检测及大数据分析...
- 【佳学基因检测】如何对多发性骨髓瘤的基因序列变化进行基因检测?...
- 【佳学基因检测】如何采用新的基因检测技术提高对多发性骨髓瘤复杂基因变化的基因检测和解析?...
- 【佳学基因检测】靶向药物基因检测如何选择使用伊马替尼治疗系统性肥大细胞增多症...
- 【佳学基因检测】定量PAI-1缺乏症基因检测Quantitative PAI-1 deficiency...
- 【佳学基因检测】F10缺陷症遗传病的基因检测多案例分析...
- 【佳学基因检测】镰刀状细胞性贫血症的基因检测...
- 【佳学基因检测】血色病基因检测、基因解码...
- 【佳学基因检测】血浆细胞计数异常基因检测...
- 【佳学基因检测】基因检测CCL22突变通过解除对微环境串扰的调控来驱动自然杀伤细胞淋巴增生性疾病...
- 【佳学基因检测】T细胞形态异常基因检测...
- 【佳学基因检测】淋巴细胞形态异常基因检测...
- 【佳学基因检测】B细胞计数异常基因检测...
- 【佳学基因检测】空泡淋巴细胞基因检测...
- 【佳学基因检测】边缘区B细胞比例降低基因检测...
- 【佳学基因检测】CD4阳性CD25阳性调节性T细胞数量升高基因检测...
- 【佳学基因检测】幼稚B细胞比例下降基因检测...
- 【佳学基因检测】CD4阳性CD25阳性,αβ调节性T细胞形态异常基因检测...
- 【佳学基因检测】自然杀伤细胞数量减少基因检测...
- 【佳学基因检测】血浆脱落比例异常基因检测...
- 【佳学基因检测】B细胞形态异常基因检测...
- 【佳学基因检测】CD4 T细胞比例降低基因检测...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
