












【佳学基因检测】骨髓增生异常综合征的基因解码与基因检测:诊断和筛查基础
骨髓增生异常综合征的基因解码与基因检测导读:
骨髓增生异常综合征 (MDS) 是一组异质的克隆性骨髓疾病,其特征是一种或多种造血谱系的不明原因的持续性外周血 (PB) 血细胞减少,或造血细胞中的骨髓 (BM) 形态发育异常,反复性基因突变,并且进展为急性髓性白血病(AML)的风险增加。在过去的几年中,随着下一代测序 (NGS) 诊断测试和新药物的发展,诊断、预后和治疗方法得到了显着改善。然而,没有单一的MDS特异性诊断参数,需要与临床信息和实验室检查结果相关才能做出诊断。
骨髓增生异常综合征的基因解码与基因检测关键词
骨髓增生异常综合征,细胞遗传学,二代测序
1. 血细胞减少症
继发性外周血血细胞减少的原因远比原发性骨髓肿瘤更常见。影响骨髓的各种非克隆性疾病,包括病毒(特别是逆转录病毒、细小病毒、肝炎病毒)、细菌和寄生虫感染、自身免疫性疾病(如幼年类风湿性关节炎、结节性多动脉炎、系统性红斑狼疮、免疫性血小板减少性紫癜)、营养缺乏症(如如营养不良、缺铁性贫血、维生素 B12 和叶酸缺乏导致的巨幼细胞性贫血、维生素 D 缺乏、锌引起的铜缺乏、维生素 A 过多症)、药物暴露、药物、毒素(如酗酒、锌、砷、铬、镉)、慢性肾脏和肝脏疾病以及内分泌疾病,首先需要基本排除,然后才能做出骨髓增生异常综合征的诊断 。诊断正确性具有实际影响,关于诊断的分歧很常见 。
血细胞减少是任何骨髓增生异常综合征诊断的“必要条件”;然而,特定的血细胞减少对分类的影响很小。此外,表现出显着形态发育异常的谱系通常与个别骨髓增生异常综合征病例中的特定血细胞减少症无关 。
根据国际预后评分系统 (IPSS) 中定义的风险分层指南,推荐的血细胞减少阈值包括血红蛋白 <10 g/dL、血小板计数 <100 × 10 9 /L 和中性粒细胞先进计数<1.8 × 10 9 /L 。然而,如果明确的形态发育异常和/ 或细胞遗传学异常存在 。除此之外,种族、年龄、性别相关差异和海拔等其他几个因素也是有价值的因素,需要考虑。
2. 形态
MDS 的形态学分类实际上取决于几种诊断方法,包括在骨髓和外周血中存在增加的原始细胞群,评估造血成分之间的细胞学异常和发育异常变化,评估细胞形态和细胞结构,以及有无纤维化。
传统上,世界卫生组织 (WHO) 根据外周血和骨髓中的爆炸百分比来定义骨髓增生异常综合征类别,但低于 20% 的阈值是强制性截止。PB 中 2-4% 或骨髓中 5-9% 的原始细胞群的存在被归类为骨髓增生异常综合征与过量原始细胞 1 (MDS-EB-1),而存在更多原始细胞,具有 5-外周血中 19% 或骨髓中 10-19%,或通过形态学检查明确存在 Auer 棒被归类为骨髓增生异常综合征与过度爆炸 2 (MDS-EB-2) 。然而,WHO 表示 20% 的原始细胞临界值并不是将患者视为患有 AML 或原始细胞转化的强制要求,并且在考虑所有信息后,治疗决定必须始终基于临床情况。出于这个原因,建议将 10-30% 的原始细胞计数纳入骨髓增生异常综合征或 AML 临床试验。
形态发育异常可能发生在一种或多种造血谱系中。红系发育异常的特征表现为巨母细胞样改变、红系前体中的多核、核分叶、核中存在固缩或染色质浓缩、涉及50%细胞膜的细胞质磨损、核间桥接、细胞质空泡化和存在环状铁粒幼细胞被描述为五个或更多颗粒,通过铁染色环绕三分之一或更多的细胞核(图1A)。另一方面,粒细胞谱系的发育异常特征可在存在成髓细胞和 Auer 棒、假 Pelger-Hüet 改变或核低分段和假 Chediak-Higashi 细胞质包涵体、异常核不规则以及异常细胞质颗粒和细胞质粒化不足(图1B)。巨核细胞发育异常的特征被描述为微小巨核细胞、小的单核、双核巨核细胞,以及存在分离的细胞核。图1C) 。应该注意的是,发育异常变化的范围可能从轻微变化到明显奇怪的异常。世界卫生组织表示,给定谱系中至少 10% 的细胞应该是发育异常的,才有资格作为一项重大发现;然而,在发育异常程度接近 10% 阈值的情况下,观察者间的变异性更成问题。这种细胞计数应在 200外周血白细胞计数和 500骨髓有核细胞计数中进行 。10% 的临界值实际上是为了防止在健康的老年人群中观察到看起来有点奇怪的造血细胞。还应该注意的是,形态发育异常在健康个体的骨髓中很常见。这就是为什么骨髓增生异常综合征诊断需要临床病史、细胞遗传学和分子研究。
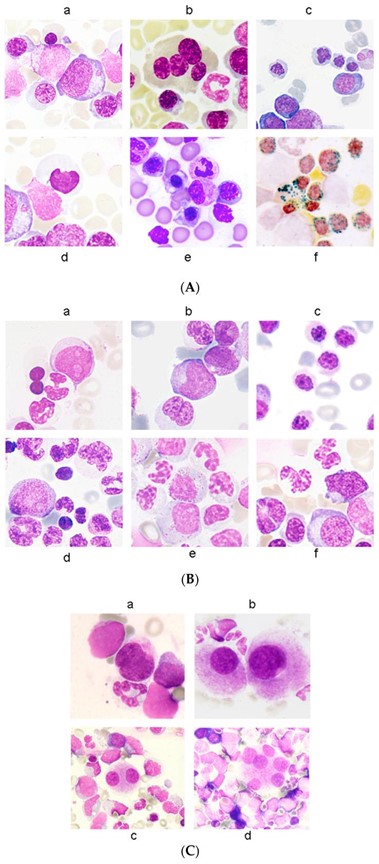
图1:红系发育不良(100×)。( a ) 巨幼细胞增多症;( b ) 多核;( c ) 核分叶;( d ) 固缩;( e ) 有缺陷的血红蛋白化和细胞质磨损;( f ) 环形铁粒幼细胞。改编自 Della Porta 等人。;(B)粒细胞发育不良(100×)。( a ) 成髓细胞;( b ) 奥尔棒;( c ) 低分叶;( d , e ) 核形状异常;( f ) 肉芽肿。改编自 Della Porta 等人。;( C) 巨核细胞发育不良 (100×)。( a ) 微型巨核细胞;( b ) 单叶巨核细胞;( c ) 小的双核巨核细胞;( d ) 具有多个分离核的巨核细胞。改编自 Della Porta 等人。。
3. 流式细胞仪
造血干细胞的改变通常被认为是骨髓增生异常综合征的疾病起始事件之一,无论是原发性新发疾病还是继发性治疗相关原因,这两者最终都会导致造血功能中断。尽管形态学被认为是诊断的关键特征之一,流式细胞术 (FC) 也被认为是诊断、预测和监测病程的重要工具 。国际和欧洲白血病网络工作组 (ELN IMDS Flow WG) 指南建议对在骨髓增生异常综合征中使用 FC 的协议进行标准化。在将细胞在一组抗体中孵育之前,应进行成熟红细胞的批量裂解方法,并获得至少 100,000 CD45 +事件是最优的。细胞发育异常特征可以反映表面抗原异常,可以使用多参数 FC (MFC) 检测到。MFC 在骨髓增生异常综合征中的应用集中在评估前体髓样抗原异常、粒细胞和单核细胞谱系中髓样成熟模式的异常。图 2A、B)、胚细胞免疫表型的计数,以及在某种程度上,祖 B 细胞(血细胞)的减少 。没有用于记录骨髓增生异常综合征的特异性 FC 标志物,但多种免疫表型异常可以确定该疾病的克隆性。与单一异常相比,多个抗原异常的存在已被证明对骨髓增生异常综合征具有更高的预测价值,因此,多个抗原表达异常的发现可能强烈支持 FC 对骨髓增生异常综合征的诊断 。抗原参数的分析可以使用四色 FC 的最低要求来实现,分析应集中在未成熟的骨髓祖细胞上。研究以下参数可以区分正常和异常祖细胞:(1)祖细胞的测量;(2) 结合前向和侧向光散射 (FSC 和 SSC) 图评估其祖细胞可塑性(CD34 和 CD45 表达);(3) CD117的表达;(4) 成熟和谱系不忠标记的表达。
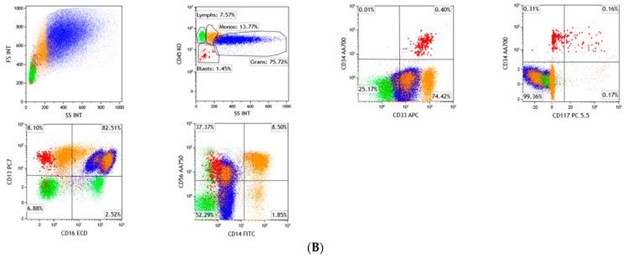
图 2:( A ) 显示有核造血细胞群的正常成熟模式。再生成髓细胞显示完整的 CD34 和 CD117 表达和异质 CD33 阳性。成熟的粒细胞前体在 CD13/CD16 图中显示出经典的“耐克旋风”模式,单核细胞显示完整的 CD14 表达,没有异常的抗原表达。绿色为淋巴细胞,橙色为单核细胞,蓝色为粒细胞,红色为原始细胞;( B )骨髓增生异常综合征病例显示 CD117 缺失和同时具有明亮 CD33 强度的成髓细胞。CD13/CD16 图中明显的异常粒细胞成熟模式,失去了经典的“耐克旋风”模式。在这种骨髓增生异常综合征病例中,粒细胞和单核细胞也表现出异常的 CD56 表达;绿色为淋巴细胞,橙色为单核细胞,蓝色为粒细胞,红色为原始细胞。
四参数评分系统(称为 Ogata 评分)被广泛用作骨髓增生异常综合征诊断的简单 FC 标准,它仅贡献最小的观察者间变异性,并且报告的特异性为 93%,敏感性为 70%。_ 该评分方案中描述的参数如下: (1) 中性粒细胞的 SSC 定义为淋巴细胞 SSC 的比值(粒细胞/淋巴细胞 SSC);(2)整个有核细胞中 CD34 +骨髓前体的百分比(CD34 +成髓细胞的百分比);(3) CD34 +前体 B 细胞 (血细胞) 在整个 CD34 +细胞中的百分比 (% of CD34 +B细胞);(4) CD34 +骨髓祖细胞的 CD45 抗原表达与淋巴细胞的比率(淋巴细胞/成髓细胞 CD45 比率)。确定了既定的参考值,与参考的偏差记为 1 分,最多 4 分。≥2 分表示 MDS,3 或 4 分的高分与骨髓增生异常综合征的高概率相关]。然而,这种评分系统不太适用于出现难治性血细胞减少症的儿科队列 。
由于红细胞生成障碍是骨髓增生异常综合征的一个共同特征,因此在流式细胞仪面板中整合红细胞异常会提高检测骨髓增生异常综合征的灵敏度。除了评估骨髓增生异常综合征中未成熟骨髓/单核细胞的抗原表达异常外,对有核红细胞免疫表型异常的分析进一步有助于支持骨髓增生异常综合征的诊断 。ELN IMDS Flow WG 已证明抗体标记物的效用,包括 CD45、CD36、CD71、CD105、CD117 和 CD235a,可用于评估红细胞发育不良,并产生评估红细胞发育不良的红细胞评分(RED 评分)异常的红系抗原表达和血红蛋白水平。IMDS 流动小组提出了红系评估指南:(1)CD36 变异系数(CV);(2) CD71 简历;(3) 平均荧光强度 (MFI);(4) CD45 阴性减少的细胞部分(祖细胞百分比)中的 CD117 阳性。CD71 增加的 CV 一直是骨髓增生异常综合征最敏感的标志物,其次是 CD117 百分比增加/减少,而据报道 CD36 增加的 CV 是最特异的标志物 。据报道,分析红系标志物 CD71 CV 和 CD36 CV 中的抗原异常,以及异常百分比的 CD117 +红系祖细胞,可提供骨髓增生异常综合征和非克隆性血细胞减少症之间的最佳区分参数。据报道,多个红系异常的存在与骨髓增生异常综合征 显着相关。
此外,对 MFC 确定的免疫表型改变的许多研究可以为未来的药物开发发现新的靶点 。
4. 细胞遗传学
细胞遗传学一直是诊断骨髓增生异常综合征的一个重要和必要的参数。WHO 严重依赖骨髓增生异常综合征中的细胞遗传学异常。除了在外周血血细胞减少症患者中建立克隆过程外,细胞遗传学在预后、临床-形态学相关性、诊断策略以及预测进展为 AML 的可能性方面发挥着重要作用。与其他髓系恶性肿瘤的诊断由单一的细胞遗传学事件(如慢性髓细胞白血病和急性早幼粒细胞白血病)不同,MDS 中有广泛的细胞遗传学定义病变,使得诊断非常具有挑战性。然而,大约 50% 的骨髓增生异常综合征具有正常的细胞遗传学。边缘发育不良和正常细胞遗传学的病例存在诊断挑战。染色体病变的各种组合促成了骨髓增生异常综合征的广泛临床病理学特征。根据世界卫生组织的说法,当顽固性血细胞减少症的所有其他继发性原因被基本且有效排除,并且只要确定了骨髓增生异常综合征定义的细胞遗传学异常(除了一些例外)。
通过常规核型分析(G-显带)和荧光原位杂交(FISH)测定的细胞遗传学研究结果,作为骨髓增生异常综合征修订后的国际预后评分系统(IPSS-R)评分中的重要参数。因此,IPSS-R 已被证明有利于预测未经治疗的骨髓增生异常综合征患者的临床结果,并有助于设计该疾病的临床试验 。综合细胞遗传学评分系统(CCSS)由世界卫生组织改编,该系统根据现有的细胞遗传学克隆定义了骨髓增生异常综合征的特定预后分层(表1)。IPSS-R 包含五个细胞遗传学亚组,它比以前的 IPSS 更重视染色体畸变(表 2)。五个细胞遗传学风险组是根据基于大型多中心数据库的新 CCSS 定义的。据报道,总生存期 (OS) 与细胞遗传学的状态存在显着差异 。
表1:骨髓增生异常综合征的综合细胞遗传学评分系统(CCSS)。改编自 Schanz 等人
|
预后亚组 |
定义细胞遗传学异常 |
|
很好 |
Y染色体缺失 |
|
缺失(11q) |
|
|
好的 |
普通的 |
|
缺失(5q) |
|
|
缺失(12p) |
|
|
删除(20q) |
|
|
双倍,包括 del(5q) |
|
|
中间的 |
缺失(7q) |
|
获得第 8 号染色体 |
|
|
19号染色体获得 |
|
|
等染色体 17q |
|
|
未在其他亚组中指定的单一或双重异常 |
|
|
两个或多个独立的非复杂克隆 |
|
|
较差的 |
7号染色体缺失 |
|
Inv(3)、t(3q) 或 del(3q) |
|
|
双倍,包括第 7 号染色体或 del(7q) 的丢失 |
|
|
复杂(三个异常) |
|
|
很穷 |
复杂(>三个异常 |
表 2:骨髓增生异常综合征的修订国际预后评分系统 (IPSS-R) 评分值。改编自格林伯格等人
|
预后变量 |
得分值 |
||||||
|
0 |
0.5 |
1 |
1.5 |
2 |
3 |
4 |
|
|
核型(CCSS) |
很好 |
- |
好的 |
- |
中间的 |
较差的 |
很穷 |
|
BM 爆炸百分比 |
≤2% |
- |
>2% 到 <5% |
- |
5–10% |
>10% |
- |
|
血红蛋白浓度 (g/dL) |
≥10 |
- |
8 到 <10 |
<8 |
- |
- |
- |
|
血小板(×10 9 /L) |
≥100 |
50 至 <100 |
<50 |
- |
- |
- |
- |
|
中性粒细胞先进计数(×10 9 /L) |
≥0.8 |
<0.8 |
- |
- |
- |
- |
- |
|
根据上述参数的总分定义五个风险组: |
|||||||
|
- 表示不适用 |
|||||||
与 AML 相比,平衡结构异常,如易位和倒位,在骨髓增生异常综合征 中相对罕见。一般而言,MDS 显示出一种特征性的、异常高的不平衡染色体异常患病率。更常见的是,这些遗传损伤往往以遗传物质染色体丢失的形式发生,例如缺失和单体性,而以三体性形式获得遗传物质的频率较低(表3)。大量病例也发生在具有复杂细胞遗传学改变(具有三个或更多异常)的患者中。
表3:MDS 中的反复性染色体异常和频率。改编自 Swerdlow 等人
|
染色体异常 |
频率 |
||
|
MDS 总体 |
治疗相关的 MDS |
||
|
不平衡 |
获得第 8 号染色体 * |
10% |
|
|
7号染色体或del(7q)缺失 |
10% |
50% |
|
|
缺失(5q) |
10% |
40% |
|
|
缺失(20q)* |
5% 至 8% |
|
|
|
Y染色体缺失* |
5% |
|
|
|
等染色体 17q 或 t(17p) |
3% 至 5% |
25% 至 30% |
|
|
13 号染色体或 del(13q) 缺失 |
3% |
|
|
|
缺失(11q) |
3% |
|
|
|
del(12p) 或 t(12p) |
3% |
|
|
|
idic(X)(q13) |
1% 至 2% |
|
|
|
均衡 |
t(11;16)(q23.3;p13.3) |
|
3% |
|
t(3:21)(q23.2;q22.1) |
|
2% |
|
|
t(1:3)(p36.3;q21.2) |
1% |
|
|
|
t(2;11)(p21;q23.3) |
1% |
|
|
|
inv(3)(q21.3;q26.2)/t(3;3)(q21.3;q26.2) |
1% |
|
|
|
t(6;9)(p23;q34.1) |
1% |
|
|
* 作为没有形态学标准的唯一细胞遗传学异常,8 号染色体增加、del(20q) 和 Y 染色体缺失不被认为是骨髓增生异常综合征的明确证据,在原因不明的持续性血细胞减少的情况下,其他异常是被认为是骨髓增生异常综合征的推定证据,即使在没有明确的形态特征的情况下。
后续样本的连续细胞遗传学分析(核型分析和 FISH)已被确定为识别骨髓增生异常综合征克隆的可能克隆进化,从而更好地了解骨髓增生异常综合征中这些遗传事件的异源性获得或丢失 。在骨髓增生异常综合征患者诊断时证明发生了克隆进化以及在较小程度上存在亚克隆,这对生存和 AML 进展具有不利影响。基线处改变的核型似乎倾向于获得进一步的细胞遗传学改变。然而,根据基线核型异常,没有出现特定的克隆进化模式。
4.1del(7q) 或单体 7
在几种髓系肿瘤中发现了 7 号单体 (-7) 和 7 号染色体长臂缺失 (del(7q)),表明其在疾病致病性中起关键作用。它们要么单独出现,要么作为复杂核型的一部分出现,并且通常与某些疾病实体的不良预后相关。在骨髓增生异常综合征中,孤立的 del(7q) 细胞遗传学异常被归类为中度预后亚组,而孤立的 -7 细胞遗传学异常被归类为预后不良亚组。在 del(7q) 患者中,与有效 -7 作为孤立异常和非复杂畸变的患者相比,存在更好的生存率。然而,由于这些相关的细胞遗传学病变之间的生存差异没有统计学意义。在大约 10% 的新发骨髓增生异常综合征病例和高达 50% 的治疗相关骨髓增生异常综合征 中报告了 7 号染色体异常。
7号染色体中的缺失断点是异质的,缺失通常是间质的。大多数病例在 7(q11) 或 7(q22) 有近端断点。在骨髓增生异常综合征中识别的 7q 上的常见缺失区域位于位置 7q22、7q32-33 和 7q35-36 。单体 7,在少数但显着数量的病例中作为唯一的细胞遗传学异常发生,可能表示涉及关键肿瘤抑制基因的主要机制 。以前的研究确定了可能导致 -7/del(7q) 发病机制的驱动基因,包括CUX1、EZH2、LUC7L2、MLL3和SAMD9/9L 。然而,尚未开发出具体的疗法。
CUX1是一种保守的、单倍体不足的肿瘤抑制因子,经常在骨髓肿瘤中缺失。它编码一个包含同源域的转录因子,位于染色体带 7q22.1。在 RNA 测序数据中,确定了与CUX1相关的细胞周期转录基因特征,表明CUX1通过调节增殖基因发挥肿瘤抑制活性 。
EZH2作为髓系恶性肿瘤的肿瘤抑制因子。它位于染色体 7q36,编码多梳族家族的成员,形成多聚体蛋白复合物,参与维持基因的转录抑制状态。它编码一种组蛋白甲基转移酶,该酶在干细胞更新相关基因的表观遗传沉默中起作用 。然而,7q 的缺失不会导致EZH2基因的丢失 。
4.2. 缺失(5q)
1974 年新颖报道了 5q- 综合征 。目前,由基因突变定义的唯一MDS亚型是在5号染色体长臂(del(5q))中具有孤立缺失的组。这种特定类型的骨髓增生异常综合征属于预后良好的亚组。最常见的症状通常是大红细胞性贫血,血小板增多症比血小板减少症更常见 。
最常见的异常包括 5 号染色体长臂的间质缺失。由于着丝粒或端粒区域的缺失或涉及NPM1或MAML1和APC基因的突变,5q 臂的更大损失与更高的风险相关MDS 和早期转化为 AML 的风险 。具有孤立性 del(5q) 的骨髓增生异常综合征是新发骨髓增生异常综合征中最常见的基因突变,它们的预后相对较好,转化为 AML 的风险降低。然而,这种异常可能是复杂细胞遗传学的一部分,其中这些病例的预后较差。从这些骨髓增生异常综合征病例中,del(5q) 不一定是主要的遗传事件,这可能是在其他疾病起始突变之后获得的,特别是表观遗传修饰突变 。
发现位于该区域的几个基因的单倍体不足能够在MDS患者中产生临床表型。例如,一个RPS14等位基因的缺失可以概括骨髓增生异常综合征del(5q) 中的红细胞生成异常。这种蛋白质的缺失已被证明会上调p53,主要是在成红细胞中,因此会促进这些细胞的凋亡。p53的突变与 del(5q) 的丢失和复杂的核型显着相关,而这与 del(7q) 无关 。大约 25% 的 Diamond-Blackfan 综合征患者也发现了RPS14的突变,这导致了RPS14的单倍体不足。常见缺失区域的其他几个基因的单倍体不足包括HSP9、CTNNA1和EGR1 。另一方面,microRNA miR-145 和 miR-146 的一个拷贝的丢失导致在骨髓增生异常综合征患者中观察到的血小板计数水平保持不变甚至增加,并伴有孤立的 del(5q) 。这些 microRNA 的丢失导致TRAF6 的上调,导致血小板增多、中性粒细胞减少和巨核细胞发育不良 。
来那度胺是一种沙利度胺类似物,对低危骨髓增生异常综合征患者显示出显着的治疗效果。在涉及染色体 5q 的间质缺失的患者中,其反应率明显较高 。它还可以减少输血需求,并可以逆转带有 del(5q) 的骨髓增生异常综合征中的细胞学和细胞遗传学异常 。
4.3. del(20q) 和 Y 的损失
20 号染色体长臂 (del(20q)) 的缺失是髓系肿瘤中反反复现的细胞遗传学异常,包括骨髓增生性肿瘤 (MPN)、MDS/MPN、MDS 和 AML。然而,与 del(5q) 不同,del(20q) 并未被 WHO 认定为骨髓增生异常综合征中的一个独特实体。Del(20q) 作为一种孤立的细胞遗传学异常,可见于无任何髓系肿瘤形态学诊断特征的患者的骨髓标本中,也可见于非髓系恶性肿瘤或不明原因血细胞减少的患者。因此,WHO 强调,在没有骨髓增生异常综合征的形态学证据的情况下,不能将孤立的 del(20q) 的存在视为不明原因的血细胞减少患者骨髓增生异常综合征的明确克隆证据。这可能会给几个骨髓增生异常综合征病例带来诊断难题。没有突变的孤立 del(20q) 患者的诊断样本进展为髓系肿瘤的风险非常低,大约三分之一的突变患者最终进展为髓系肿瘤 。
具有孤立 del(20q) 的骨髓增生异常综合征被 IPSS-R 归类为良好的细胞遗传学预后亚组。del(20q) 的断点是异构的。最常见的突变基因包括U2AF1、ASXL1、SF3B1、TP53和SRSF2。在骨髓增生异常综合征中,del(20q) 可能导致ASXL1基因的缺失,而ASXL1的改变对带有 del(5q) 的骨髓增生异常综合征产生负面影响。这也与较低的血小板计数和对氮杂胞苷 (AZA) 的不良反应有关 。ASXL1突变存在于 11% 至 21% 的骨髓增生异常综合征患者中,是 OS 差的预测因子。del(20q) 患者的中位生存期为 54 个月,而 del(20q) 加其他染色体异常患者的中位生存期为 12 个月 。
Y染色体(-Y)的缺失属于MDS中非常好的预后亚组,由IPSS-R分类,但也归因于与年龄相关的现象。值得注意的是,尽管它与骨髓增生异常综合征的更好预后有关,但确切的机制仍然未知。众所周知,女性 X 的损失和男性的 Y 损失随着年龄的增长而增加 。在大约 4% 到 10% 的男性患者中观察到 Y 染色体缺失是骨髓增生异常综合征中的单一细胞遗传学异常 。虽然 -Y 影响骨髓增生异常综合征的预后,但它发生在没有血液病证据的老年男性中。很明显,携带 -Y 的 CD34+ 细胞在男性骨髓增生异常综合征患者中比健康患者更普遍 。
4.4. 8三体
8 三体 (+8) 是骨髓增生异常综合征中最常见的染色体增加,在约 11% 的新发骨髓增生异常综合征中出现异常核型 。它属于IPSS-R分类的中间预后亚组。尽管它是一种常见的细胞遗传学异常,但在没有最小发育异常形态学标准的情况下,孤立的 +8 的存在不被视为骨髓增生异常综合征的推定证据。主要原因之一是 +8 可以作为健康个体的体质嵌合体 。几份报告表明 +8 的存在在 15% 至 20% 的骨髓增生异常综合征和急性白血病患者中是体质的。+8 的唯一存在不是骨髓增生异常综合征定义事件的另一个原因是,它也被视为再生障碍性贫血的克隆异常,这也可能是骨髓增生异常综合征的密切鉴别诊断,但在免疫抑制治疗后消失 。因此,需要存在明确的形态发育异常来区分发育不全的骨髓增生异常综合征和再生障碍性贫血。还有人指出,+8 的骨髓增生异常综合征对免疫抑制治疗反应良好,反应率高达 67% 。
4.5. 删除(11q)和删除(12p)
具有孤立性 del(11q) 的骨髓增生异常综合征与以 IPSS-R 为特征的非常好的预后(类似于 -Y)相关。它是一种罕见的细胞遗传学异常,据报道在新发和继发性 AML 和骨髓增生异常综合征中发生率为 0.7% 。KMT2A基因(以前称为MLL或混合谱系白血病基因)位于 11q23 断点处。结果表明,del(11q) 在分子水平上是异质的,可能表示涉及 11 号染色体或KMT2A基因的神秘重排。与带有神秘KMT2A的 del(11q) 的 AML 不同重排,据报道带有 del(7q) 的骨髓增生异常综合征缺乏这种神秘的重排,因此可能解释 AML 和带有 del(11q) 的骨髓增生异常综合征之间的生物学差异 。
另一个位于染色体 11q23 端粒的KMT2A基因是CBL基因。作为一种信号转导基因,由于活化的酪氨酸激酶降解受损,CBL基因的突变构成了与 AML 进展相关的重要致病病变。
12 号染色体短臂缺失是骨髓增生异常综合征的罕见事件,在诊断时发生在 0.6% 至 5% 的病例中 。这被归类为 IPSS-R 中预后良好的亚组,OS 为 76 个月 。它通常作为 12p12.2 和 12p13.1 之间非常小的间质缺失发生,影响ETV6/TEL基因 。然而,ETV6缺失在 AML 中被认为高于骨髓增生异常综合征。
4.6. 缺失(9q)
第 9 号染色体长臂的缺失在 AML 中比在骨髓增生异常综合征或 MPN 中更常见。IPSS-R 将 del(9q) 分类为中位风险亚组,中位 OS 为 32 个月。del(9q) 的髓系肿瘤被确定为TET2突变的高患病率,当TET2是唯一异常时,这种关联更为明显,发生率为 45% 。最近的数据显示,del(9q) 已从定义骨髓增生异常综合征的细胞遗传学异常列表中删除,因为它与 t(8;21) 相关,并且在 AML 中频繁发生NPM1和双等位基因CEBPA突变 。
4.7. t(17p) 或等染色体 17q
MDS 中 17 号染色体异常的存在与不良预后特征和非常低的 OS 相关,除了等染色体 17q (i(17q)),它与 IPSS-R 的中度风险预后亚组相关。在复杂核型的背景下,也发现了患者预后不良与 17 号染色体异常之间的关联。这也与 17.13.1 的丢失有关,其中包含肿瘤抑制基因 p53 ( TP53 ) 的基因座。这种细胞遗传学异常的意义在具有TP53突变的骨髓增生异常综合征和 AML 中很有价值,因为它对低甲基化剂 (HMA),尤其是地西他滨有良好的反应 。TP53是人类癌症中最常见的突变基因。它作为细胞周期停滞、DNA 修复机制、细胞凋亡诱导和细胞分化的转录因子发挥作用。在骨髓增生异常综合征中,TP53突变与 del(5q) 综合征显着相关,其在细胞周期、DNA 修复和细胞凋亡中的不同作用导致染色体不稳定和 AML 转化 。TP53在骨髓增生异常综合征发病机制中的关联也见于治疗相关骨髓增生异常综合征(t-MDS),如 2016 年 WHO 的分类所定义,其中因先前无关的恶性肿瘤或自身免疫而接受细胞毒性或放射治疗疾病被记录 。
4.8. t(11;16)
11 号和 16 号染色体的平衡易位发生在大约 3% 的治疗相关骨髓增生异常综合征病例中 。KMT2A基因(以前称为MLL基因)已被定位在 11q23 基因座中,该基因与 70 多个易位伴侣基因形成融合转录本。KMT2A基因易位导致在其氨基末端形成嵌合蛋白,并在融合伴侣基因的羧基末端部分融合 。另一方面,CBP基因在 16p13 基因座编码转录衔接子/辅激活蛋白,并参与细胞周期的调节。据推测,对 t(11;16) 阳性骨髓增生异常综合征的白血病发生的一种可能解释是,当与KMT2A融合时, CBP失去了通过其结构改变来调节细胞周期的功能。在一项研究中,t(11;16) 成年患者的 OS 与治疗相关髓系肿瘤和复杂核型的成年患者相似 。
4.9. inv(3) 或 t(3;3)
在大约 1% 的病例中观察到具有 3 号倒位 (inv(3)) 和 3 号染色体平衡易位 (t(3;3)) 的 MDS,并被归类为预后不良亚组 。inv(3)/t(3;3) 的髓系肿瘤通常表现为贫血,血小板计数可能正常或增加 。染色体畸变涉及3q26.2.2处的原癌基因 EVI1 或较长形式的MECOM和RPN1,导致 EVI1 或 MECOM 或 RPN1/EVI1 融合转录物的异位和过表达, RPN1作为EVI1的增强子表达式。EVI1与多种信号通路相关,导致细胞生长、细胞分化障碍和细胞存活 。GATA2也有牵连,并在这些情况下被观察到过度表达,表明它在染色体 3 重排的发展中的作用 。
4.10 t(6;9)
6 号和 9 号染色体易位 (t(6;9)) 在骨髓增生异常综合征中很少发生,发生在所有骨髓增生异常综合征病例的 1% 。易位导致在 der(6)中形成嵌合融合蛋白DEK/NUP214 。这种细胞遗传学事件与髓系肿瘤预后不良有关。这种异常主要作为唯一的核型畸变发生,但一个子集与复杂的核型有关 。在 AML 中, t(6;9) 患者中FLT3-ITD突变的发生率很高。有人提出,具有 t(6;9) 的骨髓增生异常综合征确实与具有 t(6;9) 的 AML 具有一些临床病理学特征,包括相对较低的血红蛋白水平、存在多系发育不良和一些突变情况,然而,它也建议骨髓增生异常综合征病例在预后上不等同于 AML 。
5. 下一代测序 (NGS)
在过去的十年中,随着高通量测序研究的发展,已确定骨髓增生异常综合征中已识别分子特征的爆炸式增长。这些重大的技术进步和遗传学突破揭开了之前隐藏的疾病驱动之谜,导致了巨大的范式转变,不仅在疾病分类方面,而且在髓系恶性肿瘤的预后和治疗指南方面。在最新的 WHO 分类中,MDS 的克隆证据主要集中在细胞遗传学改变上。然而,大约 40% 到 50% 的骨髓增生异常综合征病例具有正常的细胞遗传学,给诊断医生留下了诊断挑战。不确定潜能克隆造血 (CHIP) 的发现使情况变得复杂,。然而,CHIP 突变的存在不仅增加了进展为原发性血液肿瘤的风险。图 3A,B),但也适用于其他非造血系统疾病。在存在 CHIP 的情况下,发生血液系统恶性肿瘤的风险仅为每年 0.5% 至 1%,因此绝大多数 CHIP 患者从未发展为明显的血液系统疾病 。
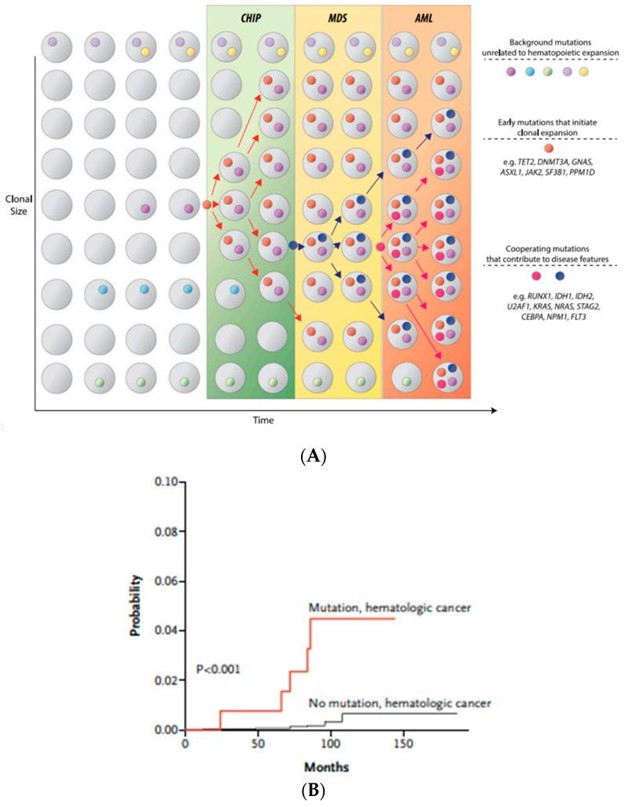
图 3:( A ) CHIP 作为血液肿瘤的前兆。体细胞突变的顺序获得导致克隆不稳定性和突变细胞的存活优势导致肿瘤克隆的扩增。改编自 Steensma 等人。;( B ) 血液系统恶性肿瘤的累积发病率。随着衰老过程中获得的体细胞突变(CHIP)数量的增加,发生血液系统恶性肿瘤的风险成比例增加。改编自 Jaiswal 等人。。
除了 CHIP,其他实体也被描述为潜在的骨髓增生异常综合征前条件。文献中不断报道了一系列临床病理和分子实体,并已在日常临床实践中进行了调整。意义不明的特发性血细胞减少症 (ICUS) 定义为在一个或多个谱系中存在持续性和无法解释的外周血细胞减少症,没有骨髓增生异常综合征相关突变的记录证据,但未能证明明显的细胞发育不良和骨髓原始细胞计数 < 5%。根据受影响的特定血细胞减少谱系,ICUS 也分为四个亚组。已经使用了诸如ICUS-A(贫血)、ICUS-N(中性粒细胞减少)、ICUS-T(血小板减少)和ICUS-PAN(双/全血细胞减少)等实体。只有在有效排除了所有潜在的鉴别诊断后,才应做出ICUS的诊断。一旦在外周血或骨髓中检测到一个或多个骨髓增生异常综合征相关的体细胞突变,如果明显骨髓增生异常综合征的诊断不足,则应诊断为意义未明的克隆性细胞减少症 (CCUS)。这意味着 CCUS 被定义为存在难治性血细胞减少,与可检测到的骨髓增生异常综合征相关突变相关,但不明显的形态发育异常。同样重要的是要认识到,与 ICUS 相比,CCUS 患者发生明显髓系肿瘤的可能性显着增加。在此范围内描述并在临床实践中采用的另一种情况是意义不明的特发性发育不良 (IDUS)。这种情况被定义为在骨髓、红细胞和/或巨核细胞中存在约 ≥10% 的显着形态发育异常,而没有报告外周血细胞减少病史,因此也不符合骨髓增生异常综合征诊断。只有在排除了反应性继发性和全身性慢性疾病和非克隆实体(例如维生素/营养缺乏、药物毒性和感染等)后,才能诊断 IDUS 。
已经证明,大约 10% 的 70-80 岁的健康个体携带一种或多种体细胞变异体,表明存在 CHIP。特定变体,特别是DNMT3A、TET2和ASXL1基因(DTA基因)中的变体,在很大程度上与 CHIP 相关,尤其是变异等位基因频率 (VAF) ≥2%。除了DTA突变外,其他基因也与 CHIP 突变有关,例如RUNX1、IDH1/2和JAK2,其中。相反,已经暗示 AML 中的某些突变谱可能来自潜在的骨髓增生异常综合征克隆,或在白血病治疗 (t-AML) 之后,特别是剪接基因突变 ( SRSF2、SF3B1、 U2AF1 和 ZRSR2 ) 已被证实对继发性 AML (s-AML) 的诊断具有 95% 以上的特异性。
对复杂的生物学和基因序列的日益增长的兴趣和理解极大地改变了骨髓增生异常综合征的诊断和治疗。尽管没有对骨髓增生异常综合征的诊断有效特异和敏感的分子特征,虽然 CHIP 中观察到的几个突变也可以诊断 MDS,但其中许多基因突变在骨髓增生异常综合征中具有显着的预测和预后价值。DNMT3A突变存在于大约 15%的MDS 病例中,它的存在与新发骨髓增生异常综合征中较短的 OS 以及发展为 sAML 的较高趋势有关]。TET2是另一种反反复生突变的基因,特别是在骨髓增生异常综合征病理生物学的早期事件中发现,据报道发生在大约 20-30% 的病例中 。TET2基因的突变也提供了重要的治疗价值。据报道,与TET2野生型相比,使用 AZA 和地西他滨 (DEC) 等 HMA 的反应率在骨髓增生异常综合征患者子集中具有更高的TET2突变,尤其是在确定主要疾病克隆时 。然而,尽管具有治疗意义,但 OS 没有显着相关性 。ASXL1 _突变是另一种表观遗传修饰因子,在大约 10-20% 的骨髓增生异常综合征中发现,移码突变和杂合点突变分别在大约 70% 和 30% 中发现,这两者都与不良预后结果和较短的 OS 相关。IDH1和IDH2见于 5-12% 的骨髓增生异常综合征患者,它们也被认为是疾病进展的早期驱动因素 。IDH变异的发生频率相对较低,表明它较少参与祖先的骨髓肿瘤克隆 。结果发现,MDS 中的IDH1 /2 突变与较低的先进中性粒细胞计数、较高的骨髓原始细胞百分比、较高的血小板计数相关,并且在比较IDH1与IDH2时,存活率没有显着差异。然而,IDH突变的存在与骨髓增生异常综合征的不良预后相关 。新出现的数据显示了新型药物的疗效,包括靶向IDH1(ivosidenib)和IDH2(enasidenib)抑制剂,用于对 HMA 抑制剂无效的高危骨髓增生异常综合征患者进行细胞减灭术 。
除了上述表观遗传修饰基因外,据报道,剪接基因突变发生在几乎一半的骨髓增生异常综合征病例中,并且在细胞遗传学正常 (CN-MDS) 的病例中非常常见,并且通常代表发病机制中的早期事件 。SF3B1是骨髓增生异常综合征中报道的最常见的突变基因,发生在大约 25-35% 的患者中。它与特定的特征细胞形态相关,并与显着的良好预后和对 luspatercept 的高反应率相关 。
还报道了一系列其他分子途径,它们对骨髓增生异常综合征的临床行为和疾病进展具有显着的生物学影响。转录因子(如BCOR、KMT2a、RUNX1、WT1和CREBBP)、信号基因突变(如KRAS、NRAS和CBL )和修复机制途径(如TP53)都已被描述。
在骨髓增生异常综合征中至少鉴定出一到两个体细胞突变(图 4) 。 目前,SF3B1基因的体细胞突变已被世界卫生组织纳入对骨髓增生异常综合征病例的一个子集的分层,该子集已被归类为具有良好的预后并与环状铁粒幼细胞的存在有关。具有环状铁粒幼细胞 (MDS-RS) 的骨髓增生异常综合征病例传统上需要来自整个红系前体的 15% 的环状铁粒幼细胞;然而,在存在SF3B1突变的情况下,截止要求下降到仅 5% 。发现MDS 中SF3B1突变的特异性为 0.97,无论是作为孤立突变还是存在其他共突变 。
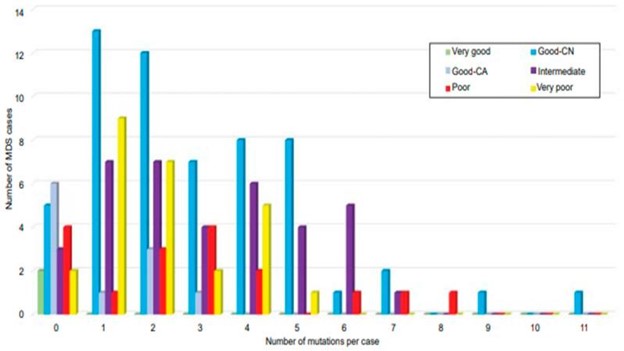
图 4:细胞遗传学预后分层MDS中共突变数量的相关性。在 69% 的细胞遗传学正常 MDS(蓝色)中发现了两个或多个突变,发生在单个患者的多达 11 个突变基因中。改编自 Tria 等人。
随着获得性体细胞突变数量的增加,MDS 患者的预后逐渐恶化。在单变量分析中,许多这些体细胞突变与预测 OS 较低有关,特别是TP53、EZH2、ETV6、RUNX1、ASXL1和SRSF2,而 SF3B1 的存在与更好的结果有关。除了SF3B1(与更好的结果相关)和TP53(与更差的结果相关),没有其他定义的变体与具有独立的预后价值有关。SRSF2和U2AF1突变的存在,或两者的共同突变,在存在骨髓增生异常的情况下显示 100% 的骨髓肿瘤特异性 。
然而,目前其他突变(除了SF3B1)尚未被世界卫生组织具体纳入作为克隆性证据或预测骨髓增生异常综合征结果的证据。
在所有继发性病因不明的血细胞减少病例中,都需要进行强制性的形态学和细胞遗传学检查。尽管如此,NGS 在这些患者队列中识别驱动突变特征肯定有助于确定克隆性,特别是在那些形态发育异常和细胞遗传学正常的患者中 。除了骨髓增生异常综合征中特定的反复基因突变外,具有较高 VAF 的变异比 CHIP 更支持骨髓增生异常综合征诊断 。对于骨髓增生异常综合征和 AML 的临界病例,原始细胞计数接近 20% 临界值,如果某些分子发现,例如NPM1、FLT3和CEBPA中的突变,NGS 可以进一步支持 AML 诊断基因,被检测出来。NGS 的发现还有助于诊断伴有外周血细胞减少的骨髓细胞减少病例,并有助于区分再生障碍性贫血和发育不全的 MDS。
反复性细胞遗传学异常与血细胞减少症患者的骨髓增生异常综合征相关,有或没有明显的形态发育异常(除了涉及 8 号染色体增加、del(20q) 和 Y 染色体缺失的唯一异常)(表3)。在具有长期和难治性血细胞减少和正常细胞遗传学 (CN-MDS) 且临床高度怀疑骨髓增生异常综合征的患者子集中,91% 的 CN-MDS 表现出潜在的突变特征,这显然可以提供髓系恶性肿瘤的克隆证据和确认骨髓增生异常综合征诊断 。最近的数据表明,NGS 不仅可以帮助证明骨髓增生异常综合征中血细胞减少患者的克隆事件,还可以预测和帮助预测 CN-MDS 病例。数据显示,与组蛋白修饰和信号转导基因野生型相反,64% 的具有组蛋白修饰突变和信号转导基因突变的 CN-MDS 患者在相对较早的时间点与 AML 转化相关。图 5A,B),因此清楚地表明可以通过 NGS 证明的突变情况应包含在 WHO 对骨髓增生异常综合征的定义中 。

图 5:(A,B)数据显示,在初始诊断时获得组蛋白修饰基因和信号基因突变的患者发生 AML 转化的速度明显更快。改编自 Tria 等人。
传统观点认为诊断时存在较高的 VAF 水平与骨髓增生异常综合征或 AML 中早期获得的体细胞克隆有关,因此 CHIP 突变已被证明是这些髓系肿瘤发病机制中最早的事件之一。这个过程可能导致进一步的遗传不稳定性,并使肿瘤更容易获得额外的突变,直到获得恶性驱动克隆。图 3A)。在发病后期获得的显性突变克隆通常与 AML 转化有关。在疾病谱的过程中,还可以通过对这些病例的连续 NGS 随访来跟踪和观察克隆进化。
全外显子组测序研究揭示了骨髓增生异常综合征中复杂而新颖的途径。剪接体机制的频繁突变也被确定为骨髓增生异常综合征中最常受影响的途径之一,尤其是SF3B1、SRSF2、U2AF1和ZRSR2中的突变。
除了新的 DNA 突变之外,现在 NGS 平台还可以检测到相互平衡的基因重排。现在可以通过靶向 RNA 测序检测到定义为 AML 的反复性基因突变,包括PML-RARA、RUNX1-RUNX1T1和CBFB- MYH11 ,这可能进一步有助于区分 AML 和骨髓增生异常综合征的模棱两可的原始细胞计数截止阈值。
由于骨髓增生异常综合征和 AML 中有大量可检测的突变标志物,NGS 为可测量的残留疾病 (MRD) 监测提供了机会和杠杆作用,尤其是在存在体细胞驱动突变(如NPM1、CEBPA、RUNX1、SF3B1等)的情况下诊断。NGS 通常可以检测到低至 1% 的 VAF,因此可以与靶向定量实时 PCR (qPCR) 或数字微滴 PCR (ddPCR) 方法发挥互补作用。使用分子标识符的纠错或条形码测序可以将 NGS 的灵敏度提高到 10 -5以用于 MRD。除了这些分子 MRD 方法之外,每条信息仍应与流式细胞术、细胞形态学/组织病理学和细胞遗传学发现相关联。对于骨髓增生异常综合征病例,与单独的细胞遗传学相比,分子评估允许更正确的预后 。
对骨髓增生异常综合征中遗传学和分子改变的临床意义的认识的进步导致了更新的风险分层模型。用于对骨髓增生异常综合征患者进行分层的个性化预测模型是一种动态分层工具,它结合了临床和基因组数据来预测骨髓增生异常综合征患者的死亡风险和白血病转化,这可以使疾病升期或降期。在对骨髓增生异常综合征患者进行分层时添加分子数据可以更正确地预测疾病行为和治疗导向策略。除了临床数据外,还使用 24 个基因组来使用机器学习技术分析骨髓增生异常综合征病例。当在分层方案中大量考虑分子数据时,可以看到对 OS 和 AML 转化的显着影响。
提出了一种更新的分子国际预后评分系统 (IPSS-M) 的提案,该系统结合了临床、细胞遗传学和遗传参数,其中主要终点是无白血病生存。IPSS-M 模型是对 IPSS-R 的改进,其中变量包括血红蛋白水平、血小板计数、骨髓原始细胞计数、IPSS-R 细胞遗传学类别以及 31 个基因中是否存在突变(Bernard 等人。 ; ASH 摘要 2021)。使用该模型和临界值,重新定义了六个风险类别:非常低;低的; 中低;中高;高的; 并且非常高(图 6)。
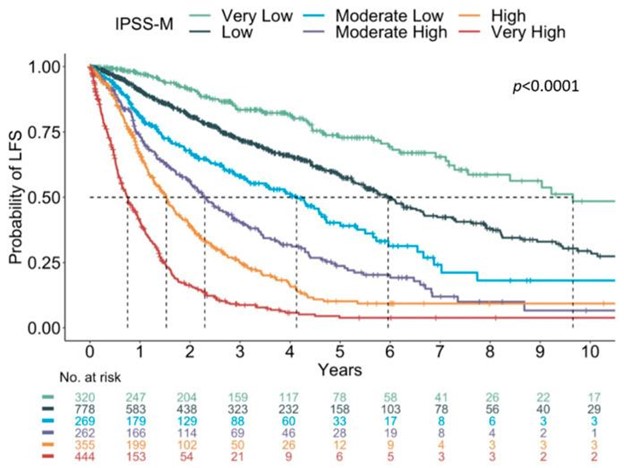
图 6:IPSS-M 中的 Kaplan-Meier 曲线显示每个亚组中无白血病存活概率的显着差异。改编自伯纳德等人
6.机器学习和未来方向
人工智能 (AI) 是一个专注于自动化通常由人类执行的智力任务的领域,而机器学习 (ML) 和深度学习 (DL) 是实现这一目标的具体方法。ML 是 AI 的一个子领域,它允许在高维空间中进行模式识别。另一方面,DL 是 ML 的一个子集,其中人工神经网络 (ANN) 用于学习复杂函数 。卷积神经网络 (CNN) 是 ANN 的一种形式,它保留了图像中像素之间的空间关系。
由于在骨髓增生异常综合征的形态学评估中存在与观察者间变异性相关的几个不可避免的问题,ML 策略可以提供一种补充工具,以尽量减少固有的主观性程度。识别骨髓发育异常以诊断骨髓增生异常综合征有时可能具有挑战性,因此 ML 方法可能有助于确定诊断。AKIRA 是第一个基于 CNN 的 AI 系统,旨在识别中性粒细胞中减少的颗粒,这是骨髓增生异常综合征中最常见的发育不良变化形式之一(图 7)。该系统报告了 97.2% 的显着高预测正确度。
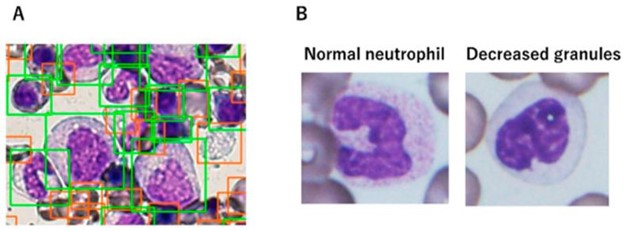
图 7:通过人工智能检测发育异常。(100×) ( A ) 检测器区分感兴趣的细胞(绿框包括所有有核细胞,红框包括红细胞、血小板和碎片);( B ) 正常成熟的中性粒细胞有足够的细胞质颗粒(左)和成熟中性粒细胞的粒细胞缺乏(右)。改编自 Mori 等人。
作为分子遗传学进展和发育不良的形态学评估的必然结果,定量图像分析提高了骨髓增生异常综合征诊断的正确性和可重复性。一种方法是使用实时变形细胞计数 (RT-DC) 成像,以标准化和测量血液或骨髓中单细胞的形态和机械特性。它利用自动图像分析和 ML 来表征数千个细胞。图像由高速摄像机捕获,并实时分析每个细胞图像,以获得细胞面积、长度和高度、纵横比、变形、惯性比和孔隙率,因此有助于细胞命名。另一个可用于诊断骨髓增生异常综合征的 ML 方法的平台是成像流式细胞术 (IFC)。IFC 将传统多参数流式细胞仪背后的技术与通过显微镜进行视觉评估的 DL 方法相结合。IFC 包含一个检测系统,可同时生成多达 12 个数字图像,这些图像是明场和荧光显微镜图像的组合。收集的数据将通过计算机算法进行分析,该算法可以量化细胞和细胞隔室的大小和形状 。
-
结论
MDS 是一种克隆性骨髓肿瘤,很难通过常规方法诊断。筛选临床和形态学评估,以及流式细胞术发现,以及通过更新的技术方法和模式检测遗传病变,特别是通过 NGS 平台进行的细胞遗传学/FISH 和突变检测,为诊断提供了一种全面的逐步方法,预后和治疗决策。总而言之,血液学家、肿瘤学家和血液病理学家应保持密切协调并不断互动,以实现整体患者护理。这种联合努力可以优化使用更新的诊断方式,以造福髓系恶性肿瘤患者及其整体福利。
更多与本文相关的科学证据,请阅读:Diagnostics (Basel). 2022 Jul; 12(7): 1581.
Published online 2022 Jun 29. doi: 10.3390/diagnostics12071581
Myelodysplastic Syndrome: Diagnosis and Screening
- 【佳学基因检测】遗传性血管性水肿基因检测案例介绍...
- 【佳学基因检测】组织细胞增生症的基因检测及其基因突变根源的介绍...
- 【佳学基因检测】血液学癌症中RNA结合蛋白的小分子抑制药物...
- 【佳学基因检测】正确理解血友病基因检测...
- 【佳学基因检测】什么人要做Diamond-Blackfan贫血(先天再生障碍性贫血)基因解码、基因检测?...
- 【佳学基因检测】NK细胞形态异常基因检测...
- 【佳学基因检测】NK细胞自然(Nature)杀伤细胞缺乏基因检测...
- 【佳学基因检测】IgA肾病易感性3型基因解码、基因检测怎么预约解读?...
- 【佳学基因检测】多发性骨髓瘤的深度基因检测及大数据分析...
- 【佳学基因检测】如何对多发性骨髓瘤的基因序列变化进行基因检测?...
- 【佳学基因检测】如何采用新的基因检测技术提高对多发性骨髓瘤复杂基因变化的基因检测和解析?...
- 【佳学基因检测】靶向药物基因检测如何选择使用伊马替尼治疗系统性肥大细胞增多症...
- 【佳学基因检测】定量PAI-1缺乏症基因检测Quantitative PAI-1 deficiency...
- 【佳学基因检测】F10缺陷症遗传病的基因检测多案例分析...
- 【佳学基因检测】镰刀状细胞性贫血症的基因检测...
- 【佳学基因检测】血色病基因检测、基因解码...
- 【佳学基因检测】血浆细胞计数异常基因检测...
- 【佳学基因检测】基因检测CCL22突变通过解除对微环境串扰的调控来驱动自然杀伤细胞淋巴增生性疾病...
- 【佳学基因检测】T细胞形态异常基因检测...
- 【佳学基因检测】淋巴细胞形态异常基因检测...
- 【佳学基因检测】B细胞计数异常基因检测...
- 【佳学基因检测】空泡淋巴细胞基因检测...
- 【佳学基因检测】边缘区B细胞比例降低基因检测...
- 【佳学基因检测】CD4阳性CD25阳性调节性T细胞数量升高基因检测...
- 【佳学基因检测】幼稚B细胞比例下降基因检测...
- 【佳学基因检测】CD4阳性CD25阳性,αβ调节性T细胞形态异常基因检测...
- 【佳学基因检测】自然杀伤细胞数量减少基因检测...
- 【佳学基因检测】血浆脱落比例异常基因检测...
- 【佳学基因检测】B细胞形态异常基因检测...
- 【佳学基因检测】CD4 T细胞比例降低基因检测...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
