












【佳学基因检测】BWS综合征的基因解码、基因检测
Beckwith–Wiedemann综合征(BWS)是一种人类基因组印记疾病,其特征是表型变异,可能包括过度生长、巨舌、腹壁缺损、新生儿低血糖、偏侧过度生长和胚胎肿瘤的易感性。标记11p15.5区域的分子缺陷可以预测家族性反复风险和胚胎肿瘤的风险(和类型)。尽管最近的知识进步,有明显的异质性在临床诊断标准和护理。正如这项共识声明所详述的,一个国际共识小组商定了72项关于BWS临床和分子诊断和管理的建议,包括从产前到成年的分子研究、护理和治疗的综合方案。一致的建议适用于Beckwith–Wiedemann谱(BWSp)患者,包括无分子诊断的经典BWS和11p15.5分子异常的BWS相关表型。尽管共识小组建议采用以分子亚组为目标的肿瘤监测方案,但根据当地医疗保健系统(例如,在美国)的不同,监测可能有所不同,应前瞻性地评估有针对性和普遍监测的结果。需要开展国际合作,包括对执行这些共识建议的结果进行前瞻性审计,以扩大最佳护理路径设计的证据基础。
Beckwith–Wiedemann综合征(BWS)是一种多系统人类基因组印记疾病,临床表现多样,分子病因复杂。BWS是一种过度生长综合征,患者通常表现为巨舌、腹壁缺损、半增生、腹部器官增大,并且在儿童早期发生胚胎肿瘤的风险增加。BWS主要是由于11p15.5区域的遗传或表观遗传缺陷引起的。这一区域包含诸如CDKN1C或IGF2等印记基因,它们是胎儿生长的强有力调节因子。虽然BWS可能出现在产前或成年期,但最常见的诊断是在新生儿期或幼儿期,估计每10340例活产中有1例患病2。自半个世纪前的第一次描述以来,佳学基因于对“Beckwith-Wiedemann”、“Wiedemann-Beckwith”或“EMG综合征”进行了搜索,获得了1500多篇BWS相关文章。然而,在BWS患者的诊断和护理方面,临床实践存在差异。
为了解决这些问题,由欧洲科学技术合作组织(COST)资助的欧洲先天性印记障碍网络发起了一个BWS共识方案,其中包括广泛的文献审查、文件草案的准备和批判性评估,以及邀请专家和专家参加的最后面对面共识会议患者组代表。这一努力产生了一系列关于诊断和护理具有新定义的贝克维思-维德曼谱(BWSp)的个体的一致建议,这些建议在本一致声明中给出。
BWS的临床方面
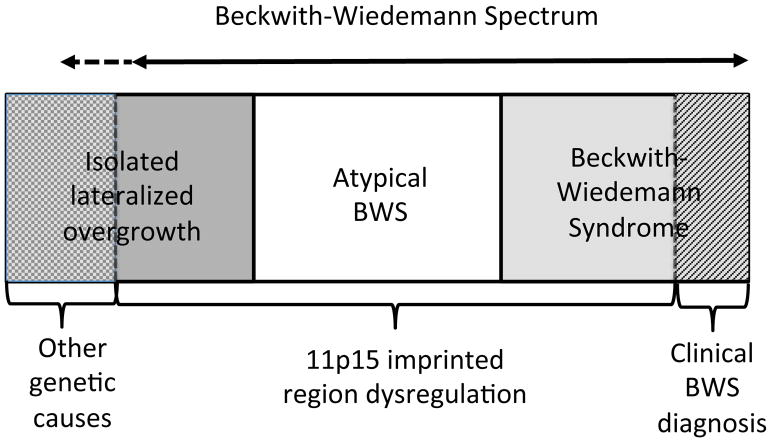
自20世纪60年代Beckwith4和Wiedemann5的新颖记载这个疾病以来,人们曾多次尝试使用各种临床标准来定义BWS,采用这些标准在不同BWS临床研究中得到不同人发病率;然而,对BWS临床标准的定义互不相同。自从20世纪90年代发现BWS患者染色体11p15.5的分子异常以来,基因解码研究人员认识到这些基因和表观遗传变化经常是镶嵌的,并导致一系列临床症状的变化。根据《人的基因序列变化与临床表征》,它包括“经典BWS”,其特征为巨舌、前腹壁缺损、产前和产后过度生长等,以及一些孤立性偏侧过度生长的病例(也有人称之为“孤立性偏侧增生症”或“孤立性偏侧增生症”);和染色体11p15.5分子异常的患者,这些患者不属于前两组,这种情况被称为“非典型BWS”。考虑到这些患者之间的有共同的临床表现和共同的分子机制,基因解码及标准制定小组决定这些表型和/或基因型组合最好被鬼归为BWSp,并且建议将其适用于BWSp患者。
| R | 建议 | 建议强度 |
|---|---|---|
| BWSp 分级是标准及BWS分子诊断的临床表现 | ||
| 1 | Beckwith–Wiedemann谱系病(BWSp)通常是由染色体11p15印记区的失调引起的,它会导致多个组织的过度生长,通常呈现镶嵌形式。BWSp包括一系列表型,儿童患者可能表现出一个或多个临床特征(见表1)。经典的Beckwith–Wiedemann综合征(BWS)和侧化过度生长(“半肥大/半增生”)被认为是BWSp的亚型(图1)。第三类被归为11p15异常的患者,他们不属于前两组。 | A+++ |
| 2 | 有许多其他的方式来诊断经典的BWS,认为巨舌,脐膨出/外淋巴管和/或(不对称)过度生长共同构成经典BWS的临床表征。尽管身高和/或体重增加(“巨大儿”)在经典BWS患者身上经常出现,它们不再被认为是BWS的主要特征。为了简洁和便于统一,基因解码制定了一致的标准(表1);经典BWS必须≥4分。符合这些标准的儿童将被视为患有BWSp,即使11p15是否异常还未确定。 | A+++ |
| 3 | BWSp中侧化过度生长是指身体一侧的长度和/或周长的大部分或全部与对侧相比显著增加,11p15异常。有11p15异常但不符合经典BWS或BWSp侧化过度生长标准的儿童仍被认为是BWSp的一部分。目前还没有足够的数据来确定侧化过度生长但没有11p15异常患者的治疗指南。还没有形成这一部分患者的诊断与治疗指南。 | A+++ |
| 4 | 建议对任何疑似BWSp的人进行分子检测(偏侧化过度生长、经典BWS或者具有表1中的特征的患者)。为简洁起见,标准建议对任何≥2分的患者进行分子诊断及基因检测。如果存在孤立性脐膨出和/或淋巴结外翻,医生可自行决定是否进行检查。当父母有可遗传的致病性11p15异常时,也建议进行检测。这种情况下,孩子有50%的患病风险。检测建议首先使用血淋巴细胞。 | A+++ |
| 5 | 低血糖是指出生后6小时内血糖水平<50毫克/dL,此后<60毫克/dL。高胰岛素血症是指葡萄糖输注速率≥8 mg/kg/min,可检测到胰岛素和/或C肽水平,不能检测到酮和游离脂肪酸。暂时性低血糖是一种提示性特征,根据上述标准定义,持续时间不到一周。这些标准将高胰岛素血症定义为持续一周以上和/或需要逐步治疗的主要特征。 | A++ |
| BWS 和辅助生殖技术 (ART) | ||
| 6 | .ART和BWS之间有着既定的联系。据估计,ART受孕个体患BWS的先进风险非常低(不超过千分之一)。需要更多的研究来进一步确定BWS与生育力降低、激素刺激、胚胎操作和印记缺陷之间的关系。 | A+++ |
BWSp的临床特点
BWS的典型特征是巨舌、巨大儿、腹壁缺损和胚胎肿瘤风险增加。佳学基因基因解码发现,并非所有的BWS患者都表现出所有这些表型特征,而且患者由于没有表现出这些特征之一而会出现漏诊、错诊。例如巨大儿,最初被认为是一个主要特征,但只有一半11p15.5分子缺陷的患者出现这种情况。作为BWSp评分系统一部分的临床特征包括,当存在时,更可能导致阳性诊断的特征(称为“主要特征”),包括巨舌、淋巴结肿大、偏侧过度生长、多灶肾母细胞瘤或肾母细胞瘤病,高胰岛素血症和特殊病理表现(如肾上腺细胞肥大或胎盘间质发育不良,表1)。巨大儿在不同的临床研究中根据具有不同的标准,这使得评估其作为主要特征的作用存在一定的问题。侧化过度生长是半肥大(或半增生)的新说法,是指部分身体的不对称过度生长。胚胎性肿瘤如肾母细胞瘤和肝母细胞瘤可发生在BWSp的诊断范围之外;然而,多灶性肾母细胞瘤更可能发生在BWSp。作为一个主要特征,高胰岛素血症被定义为持续超过一周的胰岛素水平升高和/或需要升级治疗,而暂时性低血糖在不需要进一步干预的情况下得到缓解的情况下的长期低血糖。虽然并不是总有病理结果(尤其是在产前或出生时没有怀疑BWSp,并且没有收集胎盘样本时),但在肾上腺皮质细胞肥大、胎盘间质发育不良和胰腺腺瘤的病例中,应该考虑BWSp。此外,如果有样本(尤其是出生后的胎盘),并且正在考虑诊断,病理学检查有助于作出临床诊断。
表 1:Beckwith–Wiedemann谱系征的临床特征
| 基本特征 (每条2分) | 提示性特征 (每条1分) |
|---|---|
| 巨舌 | 出生体重比平均值高出 >2 SDS |
| 脐疝 | 面部痣 |
| 侧化生成 | 羊水过多和/或胎盘肥大 |
| 多灶性和/或双侧肾母细胞瘤或肾母细胞瘤病 | 耳痕和/或凹陷 |
| 高胰岛素血症(持续一周以上,需要升级治疗) | 暂时性低血糖(持续不到一周) |
| 病理学检查:肾上腺皮质细胞肥大,胎盘间质发育不良或胰腺腺瘤病 | 典型的BWSp肿瘤(神经母细胞瘤、横纹肌肉瘤、单侧肾母细胞瘤、肝母细胞瘤、肾上腺皮质癌或棕色素细胞瘤) |
| - | 肾肿大和/或肝肿大 |
| - | 脐疝和/或直肠纵裂 |
“提示性特征”可能在一般儿科人群中独立出现,因此在下面要讲述的BWSp评分系统共识中给予的权重较小。提示性特征包括出生体重与平均体重相比大于2标准差评分(SDS)、面部红痣、羊水过多或胎盘肥大、耳皱褶或凹陷、一过性低血糖、胚胎肿瘤、肾肿大或肝肿大、脐疝或直肠纵裂。
共识评分系统及定义
曾经使用的BWS诊断标准采用了各种临床特征的组合(以巨舌、外淋巴结和/或(不对称)过度生长为主要特征),目的是优化经典和分子确诊诊断的可能性。通过分析已经发表过的临床病例,总结不同临床特征的发生率(补充表1),将这些临床表征分为主要特征或提示性特征(表1)。一些长期以来被认为是该综合征的典型特征的并不存在于每个患者身上,因此不应因为缺乏这些特征而给出不同的诊断。此外,这项共识试图包括可能是BWS的致病因素。除了告知存在典型BWS的诊断外,共识研究小组还确定,可以使用同一系统就何时进行基因检测提供指导。我们将这一新的评分系统与先前公布的系统(补充图1)进行了比较,值得注意的是先前的系统侧重于经典BWS和分子证实的BWS的诊断,而不是BWSp的诊断。
主要特征被认为是临床诊断的关键,而提示性特征增加了临床诊断的可能性和分子检测的适应症,但不太具体(表1)。在Ibrahim等人报告的BWSp研究人群中,对主要特征和提示特征进行了分析,结果表明,这些特征在很大程度上优于以前的诊断系统(补充图1)。一个局限性是,暂时性低血糖与长期高胰岛素血症在先前的队列中没有典型的区别,因此无法评估这一特征,巨大儿在先前的研究中所采用的标准并不一致。
主要特征包括巨舌、淋巴结肿大、侧化过度生长、多灶性肾母细胞瘤、长时间高胰岛素血症和BWS特有的独特病理表现。共识和以往评分系统的主要区别在于巨大儿和高胰岛素症的分类。尽管它通常与BWSp有关,巨大儿(定义为身高和/或体重>2 SDS)不再被认为是主要特征,因为它在以前的队列中定义不一,可能只出现在大约一半的BWS患者中。高胰岛素血症(定义见R5,表4)无其他可识别的分子原因可能是BWSp的最初表现特征。当高胰岛素血症持续超过一周并需要升级治疗时,被列为主要特征;当持续不足一周时,被列为提示特征。
为了简单和一致,我们使用基本特征和提示性特征制定了一致性标准(表1;R2,表4)。对于典型BWS的临床诊断,根据主要特征和提示特征,患者需要≥4分;该临床诊断不需要11p15.5异常的分子确认。评分≥2分的患者(包括评分≥4分的经典BWS患者)应根据我们的BWS调查和诊断算法进行致病基因鉴定基因解码(图3)。得分<2的患者不符合基因检测标准。基因检测阴性得分≥2分的患者应考虑进行替代诊断和/或转诊给BWS专家进行进一步评估。
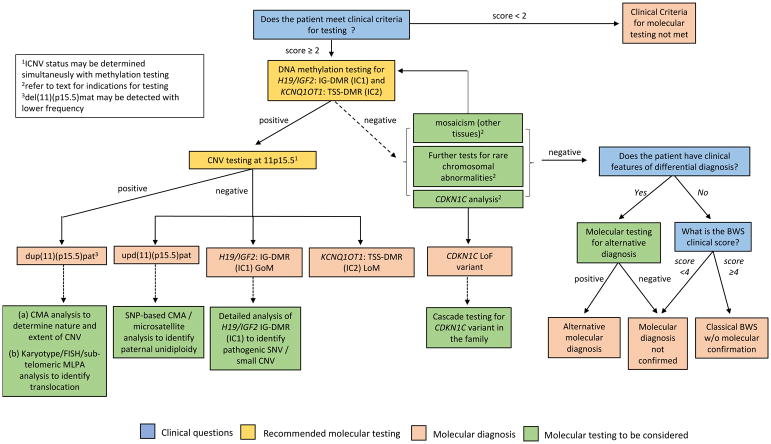
该图总结了调查疑似BWSp的分子诊断途径。临床特征得分≥2分的患者应进行基因检测。H19/IGF2:IG-DMR(IC1)和KCNQ1OT1:TSS-DMR(IC2)甲基化被推荐为一线分子检测。如果不能与DNA甲基化同时进行检测,那么在所有IC1和/或IC2甲基化异常的病例中,都应该测定DMR拷贝数。如果阳性,这些分析确定是BWSp的分子诊断,包括IC2 LOM、IC1 GOM、节段性upd(11)pat或CNV(最常见的是dup(11)(p15.5)pat)。进一步的分子测试可以用来确定甲基化异常,UPD或CNV的潜在机制。如果DNA甲基化检测为阴性,可以考虑进一步的分子检测,以确定镶嵌甲基化异常,致病性CDKN1C变异或罕见的平衡染色体重排。如果所有的分子检测均为阴性,则应考虑鉴别诊断。然而,即使没有11p15异常的分子证实,在临床评分≥4分的情况下也可诊断为经典BWS。临床问题在蓝色框中,推荐的分子检测在黄色框中,分子诊断在粉色框中,分子检测在绿色框中考虑。染色体微阵列分析,可以是基于寡核苷酸和/或单核苷酸多态性的平台。CNV,拷贝数变异;SNV,单核苷酸变异;SNP,单核苷酸多态性;LOM,甲基化缺失;GOM,甲基化获得。
1ICNV状态可与甲基化检测同时确定
2试验指示见正文
3del(11)(p15.5)mat的检测频率较低
BWSp内的临床诊断超出了经典BWS的明确诊断或明确的分子诊断是具有挑战性的,需要结合分子检测和医生意见。目前还没有足够的公开数据为评分<4且无分子异常的患者提供明确的临床建议。尽管如此,具有BWS主要特征(如巨舌症、高胰岛素症、多灶肾母细胞瘤或病理发现)的患者应转诊给具有BWS专业知识的专家进行进一步评估。与其他孤立性症状患者相比,孤立性外淋巴结患者更常见,11p15.5缺陷的可能性更小,因此不应纳入BWSp。偏侧化过度生长既可以作为BWSp的症状出现,也可以独立于BWSp9。当11p15异常出现偏侧化过度生长时,它被认为是BWSp的一部分。由于除了11p异常(例如PIK3CA、AKT1突变)外,还有多种分子原因导致偏侧过度生长,因此,不符合经典BWS标准的儿童中没有11p15异常的偏侧过度生长被认为超出了BWSp和本共识声明的范围;因此,没有提出进一步研究和临床管理的建议(R3,表4)。
BWS基因检测适应症
共识小组建议,除非有其他解释(例如,巨大儿的妊娠期糖尿病)(R4,表4),否则在得分≥2的情况下(表1)应进行分子检测。对于孤立的脐疝患者,分子检测是随意的。建议对有家族史和已知遗传致病性11p15异常的患者进行检测(10-15%的患者可能有阳性家族史)。一些先前诊断标准中包括的一些特征(例如,腭裂、骨龄晚期、多指和多乳头)暗示了另一种诊断,如辛普森-戈拉比-贝梅尔综合征,因此不包括在共识评分系统中。虽然BWSp患者的肾脏异常很常见,但通常伴有其他特征,而不是孤立的特征。当分子检测阴性时,鉴别诊断应考虑其他相关疾病(图3;补充表2)。
BWS与辅助生殖技术
辅助生殖技术(ART)被定义为在体外处理雄性和雌性配子的治疗,包括体外受精(IVF)和卵胞浆内单精子注射(ICSI)等。在工业化国家,抗逆转录病毒疗法占出生人口的1-3%。尽管这些技术被认为是安全的,但有人认为印记基因座上DNA甲基化的建立和/或维持可能受到ART的干扰。据报道,ICSI受孕的Angelman综合征分子亚型罕见,BWS患儿ART分娩频率增加(约4-6倍),一项基于人群的研究估计,IVF受孕儿童患BWS的风险约为4000分之一,大大高于一般人群。2017年的一项研究报告,抗逆转录病毒治疗可使患BWS的风险增加10倍,但先进风险约为1/1000。虽然一些流行病学研究没有发现ART后出生的儿童患BWS的相对风险增加,但分子研究支持一种关联,因为ART受孕的BWS患儿中,90%以上的着丝粒印记中心KCNQ1OT1:TSS DMR(IC2)存在外突变,相比之下,50%的BWS儿童没有通过ART40受孕。各种因素可能导致ART与Angelman综合征或BWS之间的关联,包括不孕本身(即独立于ART技术)41,42、超排或体外胚胎培养43-45。ART与表观遗传缺陷之间的联系也由大后代综合征(绵羊和奶牛中的ART相关现象,与BWS有一些表型相似)提出,据报道与BWS46中观察到的类似的表观遗传改变相关。然而,尽管有明确的证据表明ART与BWS有关,但需要借助基因解码来阐明亚生育、激素刺激、胚胎操作和印记缺陷之间的关系(表4;R6)。
BWSp的分子方面
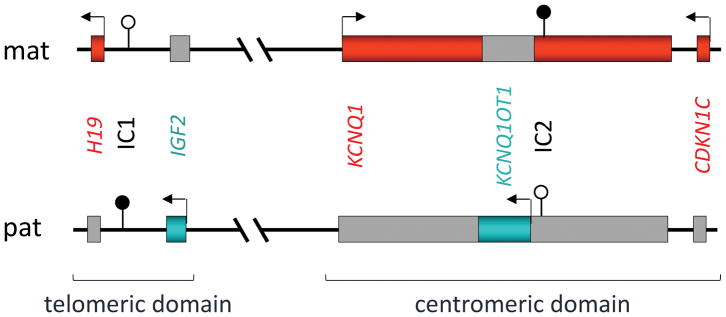
BWSp与影响染色体区域11p15.5–11p15.4内印记基因簇的分子异常有关,这些印记基因被分为两个功能独立的结构域,着丝粒和端粒结构域(图2)28。每个结构域都有自己的印记控制区,由一个差异甲基化区(DMR)标记。胰岛素样生长因子2编码基因IGF2和编码非翻译长非编码RNA(lncRNA)H19的基因位于端粒结构域,由H19/IGF2:IG DMR(H19/IGF2基因间DMR)控制,也称为印迹控制区1(ICR1)、H19-DMR或印迹中心1(IC1);OMIM*616186细胞周期抑制基因CDKN1C和编码调控性长非编码RNA KCNQ1OT1的基因位于着丝粒结构域,由KCNQ1OT1:TSS DMR(KCNQ1OT1转录起始位点DMR,也称为KvDMR、LIT1-DMR、印迹控制区2(ICR2)或印迹中心2(IC2)控制;OMIM*604115)。人类基因组变异学会(HGVS)推荐的命名法H19/IGF2:IG DMR和KCNQ1OT1:TSS DMR应在出版物和试验报告中采用47(R7,表5);不过,为简洁起见,下文使用IC1和IC2。
Beckwith-Wiedemann综合征位于染色体11p15.5
分子诊断推荐共识
| R | 建议 | 建议强度 |
|---|---|---|
| 分子遗传学分析 | ||
| 7 | 分子遗传学测试应该由在印记疾病领域有经验的健康专家进行。检测报告应采用推荐的术语(如HGV) | A+++ |
| 8 | BWSp的分子诊断应遵循图3所示的流程图。 | A+++ |
| 9 | 一线分子检测应包括H19/IGF2:IG DMR(IC1)和KCNQ1OT1:TSS DMR(IC2)的DNA甲基化分析。如果在其中一个或两个dmr处发现DNA甲基化缺陷,则应进行进一步的检测,以确定潜在的CNV或upd(11)(p15.5)pat(如果在初始诊断检测中未被鉴别)。 | A+++ |
| 10 | 鉴于与嵌合父系单亲二倍体相关的不同肿瘤谱,应考虑进一步检测以区分这种情况与upd(11)(p15.5)pat。 | A+++ |
| 11 | 在该区域存在GOM患者中,应考虑对H19/IGF2:IG DMR进行详细分析,因为SNV和/或小CNV可能发生在这些病例中,并具有高反复风险(在有阳性家族史的情况下优先考虑)。 | A+++ |
| 12 | 在甲基化检测结果为阴性的情况下,应考虑二线分子检测,可能包括CDKN1C编码外显子和外显子-内含子边界的测序(在有阳性家族史、腭裂或腹壁缺损(脐疝或外淋巴结)的情况下优先考虑) | A+++ |
| 13 | 在甲基化检测结果为阴性的情况下,应考虑二线分子检测,并可能包括分析其他组织以检测体细胞嵌合体(在存在不对称过度生长的情况下优先考虑)。 | A+++ |
| 14 | 在甲基化检测结果为阴性的情况下,应考虑二线分子检测,可能包括对罕见染色体重排的进一步检测 | A+++ |
| 15 | 如果甲基化试验结果为阴性,则应考虑二线分子检测,可能包括重新评估临床诊断和重新考虑鉴别诊断。 | A+++ |
| 16 | 遗传咨询应该由在印记缺陷领域有经验的专业人员进行。 | A+++ |
| 17 | 由于与遗传缺陷相关的反复风险(例如,CDKN1C功能丧失变异体、CNV和DMR SNV)取决于其大小、位置和父母来源,因此在为家庭提供咨询时应考虑这些因素。 | A+++ |
| 产前分子遗传学分析 | ||
| 18 | 如果产前超声检查显示BWSp的潜在特征并导致特定诊断(或排除其他潜在情况);或者如果存在已知分子缺陷的阳性家族史,这将影响相关妊娠的处理,则应考虑进行产前分子诊断调查。 | A+++ |
| 19 | 产后检查流程图(图3)不一定适用于产前检查。此流程图的修改取决于病人情况(例如,已知的分子缺陷和特定的临床特征)。 | A+++ |
| 20 | 在为BWSp提供产前诊断之前,应与父母详细讨论技术限制和伦理问题;特别是,他们应意识到,正常结果不一定排除诊断。 | A+++ |
| 21 | 建议提供产前诊断的中心前瞻性地收集关于真/假阳性/阴性诊断率的信息,并将这些信息用于多中心审计,以便进一步制定和完善最佳做法指南。 | A+++ |
约80%的BWSp26患者存在影响染色体11p15区印记基因的分子缺陷。DNA甲基化异常是最常见的缺陷;约50%的患者发现母体IC2等位基因甲基化缺失(LOM),5-10%的患者发现母体IC1等位基因甲基化增加(GOM)48。11p15.5(通常称为节段性UPD(11)pat)的嵌合节段性父系单亲等二体(UPD)可在20%的患者中检测到,5%的散发性和40%的家族性病例中检测到基因内CDKN1C突变,<5%的患者中检测到11p15的染色体异常,20%的患者未达到分子诊断28。BWS患者的孪生频率明显高于一般人群;在大多数情况下,双胞胎是女性、单卵和不协调的(即一对双胞胎受BWSp影响,另一对双胞胎不受BWSp影响)49由于发育过程中的循环共享,来自血细胞或唾液的DNA可能显示异常的DNA甲基化(通常是IC2 LOM)在受累和未受累的不协调双生子中,而在非血源性样本(如颊拭子50)中甲基化与表型一致。因此,颊拭子是不一致单卵双生子明确诊断的先进DNA来源。
BWSp的分子遗传学诊断
图3(表5;R8)给出了一个关于疑似BWSp检查的分子诊断途径的流程图。一线分子检测应测定IC1和IC2甲基化(表5,R9)。在IC2 LOM、IC1 GOM、拷贝数变异(CNV)和节段upd(11)pat(IC2LOM和IC1 GOM)51中甲基化异常。异常甲基化状态证实BWSp的诊断,但必须建立其潜在机制,以决定疾病治疗、遗传咨询和反复风险,因此,如果使用不估计DMR拷贝数的技术分析甲基化,然后,在所有IC1和/或IC2甲基化异常的情况下,应确定这一点(图3)。目前,甲基化特异性(MS)多重链接依赖探针扩增(MS-MLPA)是最常见的诊断试验,因为它同时检测DMR甲基化状态和拷贝数;然而,在低水平嵌合体的情况下,其他技术(如MS-PCR和MS qPCR)更敏感(详细清单请参阅,参见参考文件51–54)。
Recommended investigations if testing for methylation abnormalities is positive
如果使用基于PCR的方法(如MS-MLPA)暗示DMR CNV,则应考虑染色体微阵列分析(如基于寡核苷酸多态性或基于单核苷酸多态性(SNP)的阵列),以确定缺失或重复的性质和程度,以及核型分析、荧光原位杂交(FISH),或亚端粒MLPA应考虑确定可能的染色体易位。然后可以酌情将测试扩展到其他家庭成员。如果没有检测到CNV,基于SNP的阵列也将允许检测单亲二体(实际上是镶嵌)。
如果在没有CNV证据的情况下检测到IC1 GOM和IC2 LOM,则可能存在镶嵌节段upd(11)pat,如有必要,可使用微卫星分析或基于SNP的染色体微阵列分析进行确认。基于SNP的染色体微阵列被认为是研究低镶嵌的最敏感方法(例如,1–5%)分段upd(11)pat。镶嵌父系单倍性(即全基因组父系upd影响高达10%的单亲二体病例,由于镶嵌父系单倍性与额外的临床特征和肿瘤发展风险增加有关,应进行进一步的研究(SNP阵列或微卫星分析)以检测这种分子异常考虑(表5,R10)
高达20%的IC1 GOM患者可能在DMR中携带使用染色体微阵列分析无法检测到的小CNV,或八聚体结合蛋白4(OCT4)或SOX结合位点的单核苷酸变异(SNV);这些CNV和SNV与反复的高风险相关。尽管使用MS-MLPA可以检测到一些小的CNV,但是检测SNV需要额外的研究,这在大多数诊断实验室是不可用的(表5,R11)。然而,如果MS-MLPA显示IC1 GOM而无CNV,特别是有BWSp家族史的情况下,靶向IC1测序可以在专业实验室考虑。
IC2 LOM是BWSp中最常见的表观遗传学发现,但IC2 DMR缺失是罕见的,目前没有迹象表明对BWSp和IC2 LOM患者进行SNV分析。大约三分之一的IC2 LOM患者有多位点印记障碍(MLID)。在大多数患者中,MLID的临床意义是不确定的,因此通常不需要对MLID进行常规检测;然而,对于有IC2 LOM和BWSp家族史且无IC2 DMR CNV的患者,MLID检测可能有助于确定是否应考虑进一步检测反式突变。
Recommended investigations if first-line molecular testing is negative
IC1和IC2甲基化的一线分子检测结果为阴性并不排除BWSp,原因有多种,包括低于甲基化检测极限的低水平嵌合体;CDKN1C突变;罕见的平衡染色体重排(例如倒位和/或易位);BWSp病因不明或未被发现(约20%具有BWS特征表型的患者未经分子诊断);或临床诊断不正确(表5,R12–R15)。应根据最可能的原因优先进行进一步的分子检测;例如,偏侧化过度生长的较不严重表型提示嵌合,而腹壁缺损和阳性家族史的典型BWS表型则提示潜在的CDKN1C突变。
分子缺陷的嵌合体发生在大多数散发性BWSp病例中,不同的组织可能有不同比例的受累细胞。一线诊断检测通常使用血液白细胞DNA,IC1或IC2乙基化水平可能模棱两可或在正常范围内。对颊拭子、成纤维细胞培养物或间充质来源细胞(例如,从外科切除/增生组织切除术中获得)的DNA进行分析,可提高所有镶嵌缺陷的检出率(表5,R13)。
CDKN1C突变约占BWS散发病例的5%,占家族病例的40%(在母体遗传的情况下),检测候选致病性CDKN1C变异体可进行适当的级联试验,以阐明家族性反复风险。罕见的CDKN1C突变可能发生在兄弟姐妹身上,可能是由母体生殖系嵌合体引起的。
涉及染色体区域11p15的罕见母系遗传平衡易位或倒位可能与IC2甲基化异常有关,也可能与IC2甲基化异常无关,如果一线检测为阴性,则应予以考虑(表5,R14)。最后,当分子检测呈阴性时,其他相关疾病应被视为鉴别诊断(图3;补充表2,R15)。
Multi-locus imprinting disturbance
多位点印记障碍(MLID)是指除了导致主要临床表现的病变外,还伴有DNA甲基化改变的疾病。MLID在BWSp中的患病率高于其他印记性疾病,全基因组分析显示,大约三分之一患有IC2 LOM的BWSp患者存在MLID,但在节段性upd(11)pat或IC1 GOM患者中没有。在BWSp中,MLID几乎只涉及母系而非父系种系中的甲基化位点,尽管罕见病例显示IC2和IC1的LOM(后一个发现是生长限制性疾病Silver-Russell综合征(SRS)的特征)。
罕见的BWSp MLID病例与NLRP2和NLRP5(编码NACHT、LRR和PYD结构域,包含蛋白质2(NLRP2)和NLRP5)中的双等位基因母体效应基因突变有关,因此这些基因中潜在的反式作用基因突变的可能性可能值得在遗传咨询中考虑。
可能是由于MLID中可变的甲基化改变和频繁的嵌合体,其对BWS临床表型的影响仍然不清楚,因此常规临床诊断试验不是这种共识的建议。
Recurrence risks in BWSp
BWSp的反复风险取决于所确定的任何遗传或表观遗传缺陷的遗传病因和性质,以及其父母来源,因此建议家庭咨询应由在印记障碍领域有经验的个人进行,并应考虑到检测到的异常的正确性质(表5,R16,R17)。据报道,高达10–15%的BWSp病例是家族性的,最常见的原因是CDKN1C突变、染色体11p15异常和IC1内的基因改变。在这些病例中,遗传方式为常染色体显性遗传,但反复风险取决于传播受影响等位基因的父母的性别(表2)(表5,R17)。
Table 2
Summary of BWSp molecular defect categories and recurrence risk
| Molecular defect | Frequency of molecular defect | Mosaicism observed | Risk of recurrence | Characteristic clinical features (compared with other molecular subgroups) |
|---|---|---|---|---|
| IC1 GOM | 5%48 | Yes26,54,76,78,81 | ||
| IC2 LOM | 50%48 | Yes26,54,76,78,81 | ||
| upd(11)pat | 20%48 (see also paternal uniploidy) | Yes26,54,61,76,78,81 | <1%28 | |
| Loss-of-function CDKN1C variants | 5% (40% in familial cases)48 | Usually no, but has been reported rarely83 | 50% on maternal transmission82,83 | |
| Dup(11)(p15.5)pat | ~2–4%55 | No55 | ||
| Deletions involving 11p15 | 1–5%55,98 | No55 | Dependent on extent and position of CNV, and parent of origin55 | |
| Mosaic paternal unidiploidy (Genomewide paternal UPD) | Up to 10% of upd(11)pat62–67,184 | Yes62–67,184 | Low62–67,184 | High frequency of neoplasia63,64,137,152 |
| MLID | 33% of IC2 LOM cases75–78,88 | Yes78,88–91 | Low, unless an in trans genetic variant is identified79,80 | Unclear75,76,78,89,92,93 |
致病性CDKN1C变异有50%的反复风险,如果突变是从母亲处遗传,则其临床表征可变。原则上,所有11p15 CNV和平衡易位与来源依赖表型的父母有50%的反复风险。在由于易位或倒位的不平衡分离而导致11p15父系重复的病例中,携带平衡重排的个体具有正常表型。在一些家系中,观察到BWS或SRS取决于11p15重复的父系或母系传播。父传一个重复的端粒结构域和母传一个缺失的着丝粒结构域也常导致BWSp的高反复风险。与端粒或着丝粒区域内较小CNV相关的表型和反复风险的预测可能很复杂,因为这取决于它们的大小以及涉及的基因和调控元件。
当发生在母体等位基因上时,内部IC1 CNV和SNV的反复风险高达50%,尽管在一些病例中观察到不有效外显率和可能的预期。BWSp MLID的罕见家族性病例可能由母体效应基因突变(例如,NLRP2或NLRP5突变)引起,并可能与非常高的反复风险相关。在存在其他分子缺陷的情况下,反复风险通常较低(表2)。
Prenatal molecular diagnosis
BWS的产前检测带来了特殊的挑战,因为除了分子检测的一般方面(如分子干扰的范围、镶嵌检测的挑战和检测的技术限制)之外,取样前必须考虑产前检查结果的高效性和信息价值以及涉及的伦理问题。
BWSp产前诊断的主要指征是具有已知基因改变和高反复风险的家族性病例和无家族史的病例,这些病例可能具有BWSp的特征(通常是淋巴结肿大,但也有巨大儿,半肥大,产前胎儿超声检查发现了器官肿大和羊水过多(表5,R18)。因此,产前样本的诊断检测不一定遵循与产后样本相同的流程,而是反映个体情况(表5,R19)。
虽然绒毛膜绒毛(CVS)细胞、羊水(AF)细胞或胎儿血细胞(天然和培养的)可用于分子检测,但细胞培养可能会影响甲基化模式。在CVS细胞中,11p15.5处的甲基化模式可能与胚胎组织的甲基化模式不同和/或CVS细胞可能不反映胎儿的(epi)遗传构成,因此可能出现假阳性结果。由于嵌合体,所有类型的产前检查都可能出现假阴性;因此,正常的产前检查结果不能有效排除BWSp的诊断(表5,R20)。由于最近采用了产前检查,并且面临着挑战,建议前瞻性地对病例、方法和诊断率进行多中心审计,以便不断完善最佳做法指南(表5,R21)
Care and management aspects of BWSp
In view of the complex multisystem manifestations of BWSp, the consensus group recognised the requirement for effective coordination of healthcare (TABLE 6, R22).
TABLE 6
Recommendations of the management working group
| R | Recommendation | Strength of recommendation |
|---|---|---|
| 22 | It is recommended that each patient with BWSp should have an experienced lead healthcare provider who will organise the referral to each specialist, and will coordinate care for the patient. | A+++ |
| Prenatal management | ||
| 23 | If a diagnosis of BWSp is suspected or confirmed in the prenatal period, then potential BWSp-related foetal and maternal complications (for example, foetal congenital anomalies, shoulder dystocia from macrosomia, postnatal hypoglycaemia and maternal preeclampsia) should be anticipated and appropriate clinical care should be performed. | A+++ |
| 24 | If a diagnosis of BWSp is suspected or confirmed in the prenatal period, then delivery should take place in a clinical facility where neonatal intensive care can be provided. | A+++ |
| Growth and lateralised overgrowth | ||
| 25 | Growth charts from BWSp patients are needed. | A+++ |
| 26 | Physicians should be aware of the rare possibility of final height >2 SDS above the mean. Postnatal growth and pubertal development should be monitored at least annually until the end of growth. | A++ |
| 27 | Appropriate interventions might be proposed in case of possible tall stature with the same procedures as for other patients with tall stature. | A++ |
| 28 | Monitoring of leg length discrepancy should be based on clinical examination. | A++ (LO) |
| 29 | Patients with BWSp should be monitored for leg length discrepancy at least annually during childhood and referred to a paediatric orthopaedic surgeon if present. | A+++ (LO) |
| 30 | Shoe-lifts might be indicated for LLD <2 cm. Epiphysiodesis is usually indicated for predicted LLD >2 cm. Reversible epiphysiodesis might be preferred. | A++ (LO) |
| 31 | Lengthening of the shorter normal limb should be considered only for specific cases. | A+++ (LO) |
| 32 | Surgical correction of upper limbs asymmetric overgrowth is generally not indicated. | A+++ (LO) |
| Management of macroglossia | ||
| 33 | If significant airway obstruction is suspected, a careful evaluation including sleep studies (polysomnography) and/or pulmonologist consultation and ENT consultation should be performed. | A+++ |
| 34 | Tongue reduction surgery should be considered usually after the age of 1 year if there are macroglossia-associated feeding problems, persistent drooling, speech difficulties, dental malocclusion and psychosocial problems caused by the altered appearance. | A+++ |
| 35 | Surgical intervention (adenoid tonsillectomy ± tongue reduction surgery) should be considered earlier in cases of severe airway obstruction. | A+++ |
| 36 | In cases of feeding difficulties, support from a feeding specialist and dietetics should be proposed. | A+++ |
| 37 | Tongue reduction surgery should be performed by an experienced surgical team after detailed assessment by a multidisciplinary team (including paediatric anaesthesiologist, intensive care unit, surgeon, speech therapist and orthodontist) preferably in a reference centre. | A+++ |
| 38 | The results of surgery should be carefully audited and postoperative follow-up should continue until age 16 years. | A+++ |
| Management of exomphalos | ||
| 39 | Treatment of exomphalos in the context of BWSp should be in accordance with general recommendations for the treatment of exomphalos; however, in BWSp-associated cases, attention should be paid to the risk of hypoglycaemia and the anaesthetic risks associated with severe macroglossia. | A+++ |
| Management of hypoglycaemia | ||
| 40 | Capillary blood glucose should be monitored in neonates with a clinical suspicion or confirmed diagnosis of BWSp for the first 48 hours of life. Hypoglycaemia should be defined by two consecutive (30 minutes) glucose levels <50 mg/dl (2.75 mmol/l) during the 6 first hours of life or <60 mg/dl (<3.5 mmol/l) later. In case of hypoglycemia, the newborn should be transferred to a neonatal intensive care unit. | A++ |
| 41 | A diagnostic fasting test (including measurement of glucose, insulin and ketones after 6 hours of fasting for full-term babies, and after 4 hours for preterm babies) should be performed for neonates with a suspicion of BWSp 48 hours after birth and before discharge from the nursery. | A++ |
| 42 | No specific management of hyperinsulinism and/or hypoglycaemia has been proposed in the context of BWSp and management of hyperinsulinism and/or hypoglycaemia should be performed according to general recommendations. | A++ |
| 43 | In case of severe persistent hyperinsulinism in a patient with BWSp, additional causes of hyperinsulinism should be investigated. | A+++ |
| Management of cardiac lesions | ||
| 44 | Physicians should be aware of the increased prevalence of cardiac anomalies in children with BWSp. | A++ |
| 45 | A baseline, clinical cardiovascular examination should be performed at diagnosis in all children with clinical/molecular diagnosis of BWSp. Individuals with clinically detected or suspected cardiovascular abnormalities should be referred for specialist cardiac assessment and echocardiography. | A+++ |
| 46 | Annual evaluation and electrocardiogram are recommended in patients with genomic rearrangements involving the IC2 (KCNQ1OT1:TSS DMR) region. | B+ |
| 47 | Management and follow-up of congenital cardiac lesions (for example, ventricular septal defect (VSD), et cetera.) should be as in the population without BWSp. | A+++ |
| Management of neurological features | ||
| 48 | Cognitive development should be monitored by the paediatrician. Particular attention should be paid to those with risk factors such as preterm birth, neonatal hypoglycaemia, and carriers of chromosome rearrangements or paternal genome-wide UPD. | A+++ |
| 49 | For patients with a clinical diagnosis of BWSp and a learning disability with no molecular or chromosomal anomaly, other potential diagnoses should be considered and excluded (Supplementary TABLE 2) | A+++ |
| 50 | Neurological investigations, including MRI, might be indicated only in children with neurological symptoms. | A++ |
| Management of renal complications | ||
| 51 | At diagnosis of BWSp, all patients should be screened for nephrourological malformations by clinical evaluation and ultrasound scan (USS). | A+++ |
| 52 | Physicians should be aware of the possibility of hypercalciuria, which can lead to nephrocalcinosis. | A++ |
| 53 | Patients with ultrasound scan (USS)-detected anomalies should be referred to a paediatric nephrologist and urologist for specific follow-up. | A+++ |
| 54 | For patients undergoing abdominal surveillance for tumour screening, physicians and radiologists should pay attention to the possibility of nephrocalcinosis and/or stones. | A+++ |
| 55 | For patients with BWSp, at the time of adult transition, a nephro urological evaluation (clinical examination, blood pressure and USS) should be performed. | A++ |
| BWSp and embryonal tumours | ||
| 56 | Screening should be stratified according to the genotype. | A+++ |
| 57 | Abdominal ultrasound scan (USS) for BWSp-related tumours every 3 months until the 7th birthday is recommended for all patients with BWSp, except patients with isolated IC2 LOM. | A++ |
| 58 | For patients with BWSp and upd(11)pat, abdominal ultrasound scan (USS) for Wilms tumour and hepatoblastoma every 3 months until age 7 years is recommended. | A+++ |
| 59 | For patients with BWSp and IC1 GOM (H19/IGF2:IG DMR), abdominal ultrasound scan (USS) for Wilms tumour every 3 months until age 7 years is recommended. | A+++ |
| 60 | For patients with BWSp and IC2 LOM (KCNQ1OT1:TSS DMR), no tumour surveillance is recommended. | * A/B+ |
| 61 | For patients with BWSp and a CDKN1C mutation, abdominal ultrasound scan (USS) for neuroblastoma every 3 months until age 7 years is recommended. | A+ |
| 62 | For patients with BWSp and a 11p15 duplication, abdominal ultrasound scan (USS) for Wilms tumour every 3 months until age 7 years is recommended. | A+++ |
| 63 | For patients with classical BWS without a molecular defect, abdominal ultrasound scan (USS) every 3 months until age 7 years is recommended. | A++ |
| 64 | α-fetoprotein (αFP) screening is not recommended for patients with BWSp | A+ |
| 65 | Catecholamine screening is not recommended for patients with BWSp | A+++ |
| 66 | There should be a lower threshold for investigation in cases of possible tumour-related symptoms or in response to parental concerns. | A+++ |
| 67 | Treatment of tumours in patients with BWSp might be different from treatment of patients with sporadic diseases and should be discussed with respective study groups unless specific BWSp recommendations are given in the relevant tumour treatment protocols. | A+++ |
| Late-onset complications | ||
| 68 | Individuals with BWSp should be reviewed at the age of 16–18 years to identify any complications that will require continued follow-up by adult healthcare services. | A+++ |
| 69 | Young adults with BWSp should be alerted to the availability of genetic counselling so that they can seek advice prior to starting a family. | A+++ |
| 70 | Given the paucity of data on the long-term health effects of a diagnosis of BWSp, further research should be undertaken. | A+++ |
| Psychological and counselling aspects | ||
| 71 | Health professionals caring for children and families with BWSp should take a holistic approach to care and be prepared to offer referral to specialist counselling and family support services as required. Especially, psychological evaluation and support should be offered to children and their families if required. | A+++ |
| 72 | When the clinical diagnosis is confirmed, parents should be offered A+++ the contact details of BWSp support groups. | |
Prenatal management
对于已确定有反复风险的BWSp病例(表2),一些父母可能希望考虑产前诊断。如果没有分子诊断,超声检查发现前腹壁缺损、巨舌,或者更确切地说,巨大儿、内脏肿大、羊水过多、胎盘肿大或胰腺过度生长,可能提示诊断为BWS。通过产前超声扫描(USS)可检测到的罕见表现包括胎盘间质发育不良、尿路异常、心脏缺陷、肾上腺囊肿和肿块。异常的产前生化筛查结果-例如,前三个月的游离β-人绒毛膜促性腺激素(hCG)水平升高和/或后三个月的α-胎蛋白(αFP)水平升高(与脐疝有关)-可能与胎儿的BWSp有关。在已知BWSp风险增加的妊娠中,单一异常的存在(例如,外淋巴结)可能足以作出推定诊断。
在既往无BWSp病史的妊娠中,产前检测到的BWSp特征都不是孤立的病理学特征。大约10-20%的产前诊断为孤立性脐疝的胎儿和20%的胎盘间质发育不良的胎儿将患有BWSp。由于细胞遗传学和/或染色体微阵列分析表明这两个发现,BWSp的分子分析也可以在同一样本上进行,BWSp的确认或排除可能有助于父母。对于不太具体的特征(例如,尿路异常和心脏缺陷),BWSp的检测可能取决于是否存在多个BWSp相关特征。
当BWSp的产前诊断被怀疑或确认时,个体先天性异常(例如,脐疝或心脏缺陷)的处理通常遵循基于当地惯例的标准方案。然而,巨大儿在分娩时可能会引起问题(例如,肩难产),因此在妊娠后期应仔细监测生长情况,并应作出适当的分娩安排(表6,R23和R24)。BWSp也与羊水过多和早产有关。分娩后可能出现的并发症,如新生儿低血糖、巨舌引起的呼吸阻塞、脐疝的外科修复等,应予以预见,并建立适当的监测和设施。
与胎儿BWSp诊断相关的母体并发症包括妊娠高血压(~ 2.4倍风险增加)和先兆子痫。此外,在BWSp中偶尔报告HELLP(溶血、肝酶升高和血小板减少)综合征,因此,如果怀疑胎儿BWSp,这种情况应该寻找一个比正常怀孕较低的门槛相。
Growth and lateralised overgrowth
尽管在以前的报告中,产前和产后过度生长被认为是主要特征,但只有43-65%的患者出现过度生长。与其他分子亚群相比,IC1 GOM和节段性pat(11)upd患者出生时的过度生长相对更常见。出生后的生长一般处于正常范围的上部,但通常在儿童后期会减慢,在预测成人身高时,应考虑患有BWSp的儿童和没有患有BWSp的儿童生长轨迹的差异。然而,BWSp患者的生长轨迹尚未得到很好的报道,需要特定的生长图表(表6,R25)。尽管关于最终成人身高的数据很少,但有一项研究报告称,最终成人身高高于父母的目标身高,与目标身高的平均距离为1.7±1.1 SDS),大约一半的患者>2 SDS (表6,R26)。晚期骨龄是罕见的(~3%),到目前为止,没有关于BWSp患者队列中高身材治疗的数据(表6,R27)。
在BWS的所有分子亚型中都可能发生侧化过度生长,但在CDKN1C突变的患者中很少见,并且是节段性upd(11)pat10,13患者最常见的特征。11p15区域的分子异常可能在孤立性偏侧过度生长的患者中观察到,这使得在此类病例中可以诊断BWSp 。腿长差异(LLD)可能与显著的发病率相关,并对生活质量产生负面影响。LLD的管理将取决于严重程度(表6,R28-R31)。对于LLD<2cm的鞋,可能需要使用提鞋器。在孤立性(非BWSp)LLD中,骨骺发生可能被认为是LLD差异>2cm(表6,R30)。上肢不对称过度生长的手术矫正通常不适用(表6,R32)
Management of macroglossia
90%被诊断为典型BWS的儿童有巨舌症,BWSp是儿童期最常见的巨舌症病因。尽管在一些儿童中,巨舌可能会出现自发退化(由于生长速度下降和下颌骨生长增加的共同作用),但约40%的儿童接受了手术性舌复位。最常见的手术指征是进食问题;持续性流涎;发音困难;正畸问题,包括前突和前开口咬合的发展以及门牙间距/张开;以及由于不正常的美容外观和言语、进食和发音困难而导致的心理社会困难
The enlarged tongue is usually increased in size in all three dimensions and the aim of surgery is to reduce the tongue bulk while preserving normal shape and improving function. The most common surgical approach is anterior wedge resection but a variety of other techniques have been described125,128,129. Surgical complications, although infrequent, can include postoperative oedema of the tongue and wound dehiscence.
In rare cases, respiratory problems might require surgery to be performed in the neonatal period and preoperative tracheostomy might be required125. When obstructive sleep apnoea is suspected, an airway evaluation and appropriate further investigation with polysomnography can be used for objective assessment130,131 (TABLE 6, R33). In the absence of respiratory obstruction, surgery is generally delayed until at least age 12 months (when tongue size is more stable) (TABLE 6, R34–34). If the indication for surgery is unclear, the child’s progress should be monitored to determine if indications arise in the future. Long-term follow-up studies generally show favourable results of surgery in most cases with cosmetic improvement, reduced drooling, resolution of feeding difficulties, improved speech, adequate tongue mobility and, usually, no substantial effect on taste sensation126–128. Surgery has been reported to provide good outcomes in children who are operated on at a wide variety of ages, but mainly before 2–3 years125,127.
To facilitate objective assessments and accrual of accurate long-term prognostic data, surgery should, whenever possible, be restricted to a small number of units that can offer a multidisciplinary service (including an experienced surgical team) and long-term follow up (TABLE 6, R35–R38).
Management of exomphalos
Exomphalos is a cardinal feature of BWSp (TABLE 1) and is preferentially associated with molecular defects occurring within the centromeric domain (IC2 LOM or CDKN1C mutations)10,13,16. To date, no specific recommendations have been given regarding the management of exomphalos occuring in patients with BWSp compared with isolated exomphalos in accordance with usual local practices (TABLE 6, R39).
Molecular investigations of apparently isolated exomphalos in neonates rarely detect a molecular abnormality in the absence of additional BWS features132.
Management of hypoglycaemia
Hypoglycaemia in BWSp is due to excess insulin and occurs in 30–60% of children with BWSp10,13,16. Although BWSp-related neonatal hypoglycaemia is often transient and resolves within a few days, in up to 20% of neonates it can persist beyond the first week of life and require medical treatments or even pancreatectomy in the most severe cases19.
Congenital hyperinsulinism is a rare condition with a range of causes19,133. In a cohort of 501 patients with hyperinsulinism (excluding patients with focal hyperinsulinism), ~6% had features of BWSp (most of whom had segmental upd(11)pat) and half of these patients underwent surgery due to persistent hypoglycaemia after optimal medication26.
Although low plasma glucose concentrations are common during the first 24 hours of life in all newborns, by day 3 plasma, plasma glucose concentrations in neonates are similar to those of older children, with a normal range of 3.5–5.5 mmol/l (60–100 mg/dL)134. A diagnosis of hyperinsulinism is based on evidence of increased insulin secretion and/or actions at the time of hypoglycaemia, including a detectable insulin level, suppressed levels of plasma β-hydroxybutyrate (ketones), suppressed levels of plasma free fatty acids and a glycaemic response to glucagon. Diagnosis should be made in consultation with an endocrinologist who is familiar with hyperinsulinism.
Neonates with suspected BWSp should be screened for hypoglycaemia (TABLE 6, R40 and R41) before discharge from the nursery. Neonates with confirmed hypoglycaemia should be treated to maintain a plasma glucose concentration >3.9 mmol/l (>70 mg/dL)134. Management of hyperinsulinism includes medical therapies such as diazoxide and somatostatin analogs (such as octreotide and lanreotide). Surgery (pancreatectomy) might be indicated if persistent hypoglycaemia occurs despite maximal medical therapies135. New therapies such as mechanistic target of rapamycin (mTOR) inhibitors (sirolimus) or glucagon-like peptide 1 receptor (GLP-1R) antagonists have been used in treatment of hyperinsulinism and very recently in BWSp136; however, to date, no specific management (medical or surgical) for hyperinsulinism has been evaluated in the context of BWSp (TABLE 6, R42).
Two genes that are implicated in congenital hyperinsulinism, ABCC8 and KCNJ11, map to chromosome 11p, and, although rare, some patients with BWSp might carry a heterozygous mutation in either gene26. If these genes are included in the isodisomy, then homozygosity for the mutation in disomic cells produces severe hypoglycaemia26. In cases of hypoglycaemia with hyperinsulinism and without other traits suggestive of BWSp, investigations for 11p15.5 methylation abnormalities might be considered.
Management of cardiac lesions
Congenital heart disease is more prevalent in BWS than in the general paediatric population and cardiac defects occur in up to 13–20% of patients with BWS11,16,22 (TABLE 6, R44). Minor anatomical defects (for example, cardiomegaly, patent ductus arteriosus or patent foramen ovale and interatrial or interventricular defects) require echocardiographic monitoring until usual spontaneous resolution occurs (TABLE 6, R45). More severe defects might require surgical correction, although the management will be similar to that in sporadic cases of heart disease (TABLE 6, R47).
Congenital long QT syndrome has been reported in two families with BWS harbouring an intragenic deletion and a translocation at IC2 leading to inactivation of the KCNQ1 gene, which, although very rare, is associated with a risk of sudden death (TABLE 6, R46)85,102.
Management of neurological features
Cognitive development is usually normal in patients with BWSp; however, developmental delay can be associated with prematurity, severe hypoglycaemia, unbalanced chromosome rearrangements or paternal genome-wide UPD137 (TABLE 6, R48). The differential diagnosis should be carefully considered in patients with presumptive BWS and learning disability and without a 11p15 anomaly, as some overgrowth disorders (for example, Sotos syndrome, Malan syndrome and Simpson–Golabi–Behmel syndrome) are more frequently associated with developmental delay30,138–144 (Supplementary TABLE 2) (TABLE 6, R49).
Malformations of the central nervous system (for example, abnormal posterior fossae (including Dandy–Walker malformations) or abnormal corpus callosum or septum pellucidum) have been reported in rare patients with BWSp (chiefly with a defect involving IC2)83,145, and these features might need to be considered in children with neurological symptoms or signs (TABLE 6, R50).
Management of renal complications
The prevalence of nephro-urological anomalies in BWSp is 28–61%146. A variety of anomalies have been described; cortical and medullary cysts occur in ~10% of patients with BWSp and the prevalence of hypercalciuria and nephrolithiasis is increased compared to the general population 147. Although not all nephro-urological anomalies detected by ultrasonography will be of clinical significance, a minority of anomalies might be severe (and usually detectable prenatally), requiring medical or surgical management. Severe vesicoureteral reflux might cause kidney damage and recurrent urinary tract infections148 In addition, nephromegaly might be a marker of increased risk of Wilms tumour146.
Renal anomalies might occur in all molecular subtypes of BWSp, but only certain groups might be offered regular renal imaging for tumour surveillance. Management of the nephro-urological aspects of BWSp should be pragmatic and balance the benefits of presymptomatic diagnosis and treatment of critical obstructions and urinary tract infections for preserving renal function with the drawbacks of over-investigation for benign variants detected by surveillance. Thus, we recommend a nephrourological evaluation at clinical diagnosis and at the time of adult transition for any patient with BWSp, and screening for nephrocalcinosis and/or stones only in patients who undergo abdominal USS for tumour screening (TABLE 6, R51–55).
BWSp and embryonal tumours
Embryonal tumours occur in ~8% of children with BWSp149. The most common types of embryonal tumour are Wilms tumour (52% of all tumours), hepatoblastoma (14% of all tumours), neuroblastoma (10% of all tumours), rhabdomyosarcoma (5% of all tumours) and adrenal carcinoma (3% of all tumours) 13. Although there are some differences in mean age at diagnosis between tumour types, the overall cancer risk is highest in the first two years of life and clinical experience suggests that the cancer risk then declines progressively before puberty, approaching the cancer risk of the general population. Currently, there is no evidence of an increased risk of malignant tumours in adulthood (Supplemental TABLE 3).
The tumour risk correlates with the BWSp molecular subgroup; patients with segmental upd(11)pat and IC1 GOM have a higher tumour risk than patients with CDKN1C mutations and IC2 LOM58. The four main molecular subgroups are characterised by a cancer risk gradient, with the highest risk in cases of IC1 GOM (28% risk), followed by segmental upd(11)pat (16% risk), CDKN1C mutation (6.9% risk) and IC2 LOM (2.6% risk) 13. In addition, there are also differences in the tumour types observed between molecular subgroups. Patients with IC1 GOM are mostly predisposed to developing Wilms tumour (observed in 24% of cases and accounting for 95% of malignancies in this group)13,14,149. Conversely, patients with IC2 LOM and CDKN1C mutations do not usually develop Wilms tumour, but rather develop other tumours such as hepatoblastoma, rhabdomyosarcoma and neuroblastoma. Thus, a study from 2016 reported a prevalence of Wilms tumour of ~0.2% (2/995) in patients BWSp and IC2 LOM149; Although, a report from 2017 suggested that the risk of Wilms tumour in patients with IC2 LOM might be underestimated, only a single patient with Wilms tumour and an IC2 epimutation was observed150 so that, when put together with previous reports of Wilms tumour in large cohorts of patients with BWSp, the overall prevalence of Wilms tumour with IC2 LOM is probably much less than 1%151. Patients with CDKN1C mutations are mostly predisposed to neuroblastoma13,14,149. Patients with segmental upd(11)pat are predisposed to develop any of the tumour types seen in BWSp (TABLE 3). Individuals with genome-wide paternal UPD seem to have a high risk of developing tumour types similar to those with segmental upd(11)pat, but with an increased incidence of hepatic and/or adrenal tumours extending into adolescence and young adulthood63,64,137,152.
Table 3
Proposed tumour surveillance protocol for Beckwith–Wiedemann spectrum
| Tumour risk (% of patients) | Tumour type for surveillance | Surveillance procedures | Timing | Refs column |
|---|---|---|---|---|
| IC2 LOM | ||||
|
Tumour incidence lower than other molecular subgroups; extremely variable tumour spectrum; only half of tumours arise in the abdomen |
|
13 | |
| IC1 GOM | ||||
|
Wilms tumour | Abdominal USS | Every 3 months from diagnosis until age 7 years | 13 |
| upd(11)pat | ||||
|
|
Abdominal USS | Every 3 months from diagnosis until age 7 years ^ | |
| CDKN1C mutation | ||||
|
Neuroblastoma | Abdominal USS | Every 3 months from diagnosis until age 7 years | 13 |
| Classical BWS with negative molecular tests | 13 | |||
|
Wilms tumour | Abdominal USS | Every 3 months from diagnosis until age 7 years | |
Proposed tumour surveillance protocol for patients with Beckwith–Wiedemann spectrum disorder (BWSp; including those with isolated lateralized overgrowth who have 11p15 abnormalities) are shown, stratified according molecular subtype. Although there are differences in tumour risks and prevalent tumour types between molecular subgroups when surveillance is recommended, a single surveillance programme is used to reduce confusion and enhance consistency. In specific healthcare systems, practice might currently vary from this protocol (see the text for details).
Specific studies investigating the tumour risk in patients with isolated lateralized overgrowth and clinically diagnosed BWS with negative molecular testing are lacking. It seems plausible that the cancer risk in patients with isolated lateralized overgrowth who fall within the BWSp is linked to the type of 11p15.5 molecular anomaly. Indeed, the tumour risk in patients with isolated lateralized overgrowth and segmental upd(11)pat is estimated to be as high as 32–50%121,153.
Tumour surveillance strategies
Tumour screening in patients with inherited cancer predisposition syndromes aims to improve patient survival and reduce morbidity through earlier detection of tumours. However, no surveillance protocol can detect every tumour and there are both benefits and drawbacks to screening — the latter include the financial costs, morbidity that can result from investigating asymptomatic benign lesions detected on surveillance, and psychosocial burden of repeated investigations for the patient and family. There is no generally accepted risk threshold for instigating tumour screening strategies and it might vary according to regional medical and medicolegal practices and local healthcare systems. Although screening is considered for a tumour risk >1% in the USA, a risk of 5% might be considered an appropriate threshold in Europe9,154. Various protocols have been suggested for tumour surveillance in BWSp, usually comprising abdominal USS with or without measurement of αFP levels at various ages and intervals during infancy13,14,155. Traditionally, although most protocols have been applied to all cases of BWSp, the definition of specific epigenotype-tumour risk correlations provides a basis for more targeted surveillance protocols.
Screening for Wilms tumour
Abdominal USS is the preferred modality for Wilms tumour screening. The doubling time of Wilms tumour cells, has been estimated to be 11–13 days156 and USS is recommended every 3–4 months157,158. Given the high survival rate of individuals with Wilms tumour (90% overall survival at 4 years), early detection of Wilms tumour by surveillance is predicted to only marginally impact survival; however, diagnosis at an earlier stage might reduce the burden of treatment-related morbidity159–162.
If Wilms tumour screening is targeted by BWSp molecular subgroup, patients with IC1 GOM and segmental upd(11)pat are at the highest risk and several groups have suggested that patients with IC2 LOM should not be screened using USS in order to avoid excessive medicalization and possible false-positive results149,154.
Screening for hepatoblastoma
The risk of hepatoblastoma in patients with BWS is >2000-fold higher than in the general population and hepatoblastoma is the second most common tumour type in BWS20. However, specific studies evaluating hepatoblastoma screening in BWSp are lacking. Abdominal USS is a first-line investigation in children with a suspected liver mass, although not all parts of the liver can be imaged easily and small tumours might be missed159. Concerns about the sensitivity of abdominal USS led to suggestions that it should be combined with measurements of serum levels of αFP, which is secreted by >95% of hepatoblastomas163–165. Treatment and outcome of patients with hepatoblastoma is closely connected to tumour stage at diagnosis, and preliminary data suggested that patients with BWSp and hepatoblastoma who are screened for αFP have an earlier stage at diagnosis and a better prognosis than unscreened patients, and that increased serum αFP levels might precede hepatoblastoma detection by USS166. However, this hypothesis is unproven and further data is required. In the paediatric setting, interpreting serum αFP levels can be complex due to the wide range and variable concentrations in early infancy167,168. Serum αFP levels might be higher in babies with BWS and without hepatoblastoma than in normal age-matched healthy controls169. In view of the burden of repeated venepuncture and the complexity of interpreting elevated αFP levels, it has been debated whether the benefits of αFP screening in BWSp outweigh the drawbacks170–173 and the consensus voted not to recommend αFP screening (TABLE 6, R64).
Screening for neuroblastoma
Although reported in all BWSp molecular subgroups, neuroblastomas are preferentially associated with CDKN1C mutations with a frequency of ~4%13 (TABLE 3). Detection of asymptomatic neuroblastomas by determination of the urinary tumour markers vanillylmandelic acid and homovanillic acid and/or the catecholamine to creatinine ratio, combined with three monthly USSs until age 2–3 years has been suggested149. However, previous neuroblastoma screening strategies using urinary markers in large-scale paediatric settings had a very minor influence on the related morbidity and mortality rates174,175 and there is currently no evidence that neuroblastoma screening in BWSp improves treatment and survival (TABLE 6, R65).
Surveillance for other tumour types
Screening for adrenal carcinoma can be undertaken using clinical evaluation, adrenal USS and determination of serum dihydroepiandrosterone sulfate (DHEAS) concentrations every 4–6 months 176. However, adrenal carcinoma is rare in BWS (even in patients with genome-wide paternal UPD who are at highest risk) and there is no data on the utility of such screening strategies in BWSp.
Consensus tumour surveillance protocol
共识小组一致认为,肿瘤监测应针对风险最高的BWSp分子亚型,不应向患有BWSp和IC2 LOM的儿童提供常规USS(表6,R60)(尽管针对症状或父母关注的调查应有较低的阈值)。其他BWSp分子亚群和典型BWS且未发现分子异常的患者应每3个月接受一次腹部USS治疗,直到7岁(表6,R57-59和R61-63)(表3)。一致认为,不应常规提供α-FP测量,因为判断HB的发病率太低,无法高效进行特异性筛查,对患者和家属的监测影响不明确,解释困难可能导致假阳性结果(表6,R64)。然而,在特定的医疗保健系统中,临床医生目前可能与提议的方案有所不同,特别是在区域方案可用的情况下,会根据针对性和普遍监测的前瞻性研究结果而改变。 共识监测方案使约50%的低肿瘤风险BWSp儿童免于3个月的USS,尽管在CDKN1C突变的患者中,肾母细胞瘤的风险很小,但应用了一种常见的监测模式(在某些亚组中,腹部USS而不是肾USS,其他亚组的肝脏USS等)在所有要筛选的组中避免了与更复杂方案混淆的可能性。值得注意的是,商定的方案不同于美国癌症研究协会(AACR)儿童癌症易感研讨会最近建议的方案,后者采用1%的风险阈值进行监测,因此建议对所有BWS病例进行腹部USS和αFP筛查。AACR组和这一共识组都是基于不同分子亚组中类似的肿瘤风险数据做出这些决定的,但在采用靶向筛查方法方面得出了不同的结论。应当指出的是,AACR小组主要由来自北美的专家组成,而国际BWS共识小组主要由来自欧洲中心的专家组成,这些中心已经在一些国家采用了针对性筛查。因此,这两个群体在筛选建议上的差异主要反映了北美和欧洲不同的医学和医疗规定。尽管考虑到AACR小组和专家组的不同结论,普遍同意的筛查方案通常更为可取,但目前欧洲和北美的筛查方案可能有所不同是合理的。这种做法的多样性是有帮助的,因为对这两项议定书的结果进行仔细审计,有助于在今后的国际协商一致会议上进一步完善我们的建议
BWSp相关肿瘤的治疗
患有BWSp和肾母细胞瘤的儿童与患有肾母细胞瘤的非综合征儿童相比,由于早期疾病、间变性肿瘤较少以及双侧同步或异时反复的发生率较高,因此转移性疾病较少(表6,R67)。后者似乎与一个或两个肾脏中存在多灶性或弥漫性肾原性休止(肾母细胞瘤病)有关,这一特征在标准图像上与肾母细胞瘤不易区分。虽然BWSp和肾母细胞瘤患者通过腹部USS监测诊断的肿瘤大小小于散发性肾母细胞瘤的儿童,但总体生存率相似(4岁时至少90%)。较小的肾母细胞瘤更易接受肾部分切除术,考虑到进展性非恶性肾脏疾病和双侧肾母细胞瘤的潜在并存性,BWSp患者尤其先进保留肾单位的策略(如肾部分切除术)。2016年的数据显示,BWSp和肾母细胞瘤患者在保留肾单位手术和全肾切除术后的结果具有可比性。
迟发综合症
BWSp的特征,如巨舌症和出生后过度生长,往往随着年龄的增长而改善,因此,BWSp在成人中往往被忽视,除非在儿童期有事先诊断。关于成人BWSp(R70)患者的长期预后和迟发并发症的资料很少。对与BWSp儿童期特征不直接相关的潜在成人发病并发症的关注分为四个方面。
肿瘤形成
尽管BWSp与胚胎性肿瘤有联系,但BWSp与成人常见发病癌的易感性之间没有明显的联系。虽然在BWSP的成人中报告了罕见的内分泌肿瘤(补充表3),但没有证据表明有特定的肿瘤风险可以作为增加监测的依据。然而,尚未对大量患有BWS的成人进行随访。BWSp患儿在接受胚胎性肿瘤治疗后,可能会因手术、放疗或化疗而出现迟发性并发症,与散发性肿瘤患儿相似。
心血管缺陷
先天性心脏病患者需要在成人专科诊所进行适当的随访。虽然罕见,心血管缺陷可能在成年后第一次被诊断,但常规筛查并不适用。有可能诱发长QT综合征的罕见IC2 CNV和/或重排的患者需要在整个成年期进行随访。
不孕
虽然BWSp中描述了泌尿生殖道的先天性异常(例如,双角子宫),但没有明确的证据表明BWSp妇女存在过度生育问题。据描述,受累男性(与女性相比)的生育能力降低,但BWSp男性不育的频率尚不清楚。
肾脏异常
尽管成人BWSp患者肾脏异常的诊断实例已有报道,但据推测,如果进行肾脏USS,通常会在儿童期发现肾脏异常。
尽管已建议对BWSp成人进行定期监测(例如,超声心动图、肾功能测试和听力评估),但在儿童期监测期间未发现异常,在无症状的成人BWSp患者中进行此类调查的检出率可能很低,并可能对医疗保险造成问题。共识小组一致认为,应在16岁时进行详细的临床检查和肾脏USS(补充表4),并同意仅基于持续问题继续监测的具体建议(表6,R68)。应鼓励患有BWSp的成年人在成家前寻求遗传咨询意见(表6,R69)。在这一阶段,任何有关生育率的潜在问题都可以审查,并酌情转诊作进一步调查。
心理和咨询方面
BWSp等疾病的诊断会对家庭的心理和社会福利产生广泛的影响。尽管确切的影响因家庭而异,并且会受到个别医疗和社会因素的影响,每个家庭可能面临不同的挑战,但重要的是,所有医疗专业人员都要意识到可能与家庭相关的更广泛的非医疗问题(表6,R71)。BWSp可能特有的心理社会方面的信息很少。在许多情况下,既往没有相关的家族史,父母也没有为诊断做好准备。肿瘤风险等问题令人担忧,不同的医疗实践和建议可能会导致父母的不确定和焦虑。一项针对患有巨舌症的BWSp儿童家长的调查显示,家长普遍担心,大舌头突出和持续流口水会导致陌生人盯着他们,并质疑他们的孩子是否有学习困难。家长们还担心这可能会导致其他孩子取笑,一项回顾性问卷调查显示,在BWSp儿童中,情绪困难和同伴问题明显增多。医疗保健专业人员应意识到可能会出现心理社会困难,并应准备将家庭转介给遗传咨询师、社会工作者和心理学家等专家(视情况而定)。由于支持小组在帮助家庭适应诊断、分享他们的关注和经验以及获得正确的护理和支持方面可以发挥关键作用,因此应向所有家庭提供相关小组的联系方式(表6,R72)。 总结
本共识声明中描述的第一个国际BWS共识小组的建议为改善BWSp的诊断和治疗提供了一个框架。由于BWSp具有复杂的遗传和可变的多系统表型的特点,因此为每位患者指定一名主治医师(表6,R22)以确保在整个儿童期内协调护理的许多方面是很重要的(补充表4)。建议的诊断和治疗方案旨在实用且具有成本效益(例如,与普遍的监测策略相比,针对高危人群的肿瘤监测应降低成本)。然而,在一些医疗系统和医疗法规环境中,可能需要进一步的证据来改变临床实践(例如,北美的肿瘤监测)。因此,重要的是,在执行这些协商一致建议的同时,还应进行前瞻性的安排,以便为今后的协商一致倡议扩大证据基础。
- 【佳学基因检测】基因检测揭示什么样的自闭症患者会有智力障碍?...
- 【佳学基因检测】卵巢早衰基因检测可以找到发病原因吗?...
- 【佳学基因检测】北京Y染色体微缺失基因检测...
- 【佳学基因检测】长期不育男人原因查找基因检测...
- 【佳学基因检测】孕前遗传基因检测挂什么科?...
- 【佳学基因检测】第一个胎儿流产,为什么母亲要做染色体检查基因检测?...
- 【佳学基因检测】婚前基因匹配和新生儿出生前基因筛查检测哪一个更好?...
- 【佳学基因检测】疑似女性不育症为什么要做X染色体基因检测?...
- 【佳学基因检测】女性不育症的基因检测分析...
- 【佳学基因检测】基因解码鉴定与女性不育症有关的减数分裂基因...
- 【佳学基因检测】女性不育症基因检测需要排除反复流产和胎儿死亡因素...
- 【佳学基因检测】受精后形成葡萄胎是女性不孕症不可忽视的原因之一...
- 【佳学基因检测】将母体效应基因与受精后植入失败基因纳入女方不孕症的基因检测分析中...
- 【佳学基因检测】女性生殖障碍基因解码揭示授精基因突变是女性不育症的一个原因...
- 【佳学基因检测】注意:排除女方不育原因的基因检测要包括多囊卵巢基因突变!...
- 【佳学基因检测】解码激素作用途径,实现女方不孕原因基因检测的完整性...
- 【佳学基因检测】女性不孕原因查找为什么要包括线粒基因序列检测?...
- 【佳学基因检测】不孕不育基因检测为什么要包括导致导致原发性卵巢功能减退症的基因突变?...
- 【佳学基因基因检测】基因解码揭示部分女性不孕症是卵泡生成相关的基因缺陷引起的...
- 【佳学基因检测】女性不育症基因检测为什么必须包括DNA修复基因?...
- 【佳学基因检测】早期卵细胞发生和染色体分离缺陷导致女性不孕症...
- 【佳学基因检测】女性不育症的性别发育不全基因...
- 【佳学基因检测】来曲唑与女性不育症基因检测...
- 【佳学基因检测】多囊卵巢综合征遗传吗,怎么阻断?...
- 【佳学基因检测】多囊卵巢综合征是遗传性疾病吗,基因检测靠谱吗,怎样才能治好呢?...
- 【佳学基因检测】多囊卵巢综合征(PCOS)多样本基因检测的数据揭示了什么?...
- 【佳学基因检测】个性化健康 DNA 检测:健康投资...
- 【佳学基因检测】基因检测:它是什么、它是如何工作的以及它的用途?...
- 【佳学基因检测】试管婴儿胚胎植入前为什么要检测癌症基因?...
- 【佳学基因检测】ugt1a1基因检测意义...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
