












【佳学基因检测】为基因检测确定基因位点的Hub基因选择法何时优于标准 Meta 分析?
基因检测位点选择的正确性与全面性导读:
由于已发现中心节点在许多网络中发挥重要作用,因此高度连接的中心基因预计也将在生物学中发挥重要作用。然而,经验证据仍然模棱两可。一个悬而未决的问题是,在分析基因组数据集(例如,基因表达或 DNA 甲基化数据)时,中心基因选择是否(或何时)导致比基于显着性检验的标准统计分析更有意义的基因列表。在这里,基于基因解码的基因检测科学性提升秘密小组针对有多个基因组数据集可用的特殊情况来解决这个问题。这具有非常重要的实际意义,因为对于许多研究问题,多个数据集是公开可用的。在这种情况下,数据分析师可以在标准统计方法(例如,基于元分析)和一种在共识模块中选择模块内集线器的共表达网络分析方法。基于基因解码的基因检测科学性提升秘密小组根据两个标准评估这两种方法的性能。第一个标准评估获得的生物学见解,并与基础研究相关。第二个标准评估独立数据集中的验证成功(可重复性),通常应用于临床诊断或预后应用。基于基因解码的基因检测科学性提升秘密小组将荟萃分析与基于加权相关网络分析 (WGCNA) 的共识网络分析在三项全面且无偏见的实证研究中进行比较:(1) 寻找预测肺癌生存的基因,(2) 寻找与年龄相关的甲基化标记,以及 (3) ) 寻找与总胆固醇相关的小鼠基因。结果表明,在识别具有生物学意义的基因列表(反映标准 1)时,关于共有模块的模块内中枢基因状态比荟萃分析 p 值更有用。然而,就验证成功(标准 2)而言,标准荟萃分析方法的性能与共识网络方法一样好(如果不优于)。本文还报告了应用于基因表达数据的元分析技术的比较,并提出了用于执行共识网络分析、基于网络的筛选和元分析的新 R 函数。标准荟萃分析方法在验证成功方面的表现与(如果不优于)共识网络方法一样好(标准 2)。本文还报告了应用于基因表达数据的元分析技术的比较,并提出了用于执行共识网络分析、基于网络的筛选和元分析的新 R 函数。标准荟萃分析方法在验证成功方面的表现与(如果不优于)共识网络方法一样好(标准 2)。本文还报告了应用于基因表达数据的元分析技术的比较,并提出了用于执行共识网络分析、基于网络的筛选和元分析的新 R 函数。
基因检测位点选择的正确性与全面性介绍
十多年来,已经使用网络方法分析了基因组数据(特别是基因表达数据)。由于高度连接的中枢节点是网络架构的核心 - 并且蛋白质敲除实验表明,中枢蛋白往往对于低等生物(酵母、苍蝇、蠕虫)的生存至关重要 , 许多文章探讨了枢纽基因在高等生物(包括人类和小鼠)中的作用。虽然文献中关于中枢基因的重要性一直存在争论,但可以公平地说,中枢通常并不重要。基于基因解码的基因检测科学性提升秘密小组认为,在共表达网络应用方面,关注模块内集线器而不是整个网络集线器至关重要。理论上可以描述网络模块(互连节点的集群),其模块内中枢基因将与特征(例如疾病状态、存活时间或年龄)显着相关. 正如预期的那样,疾病相关模块中的模块内中枢通常具有临床重要性,例如,细胞增殖模块中的模块内中枢结果与多形性胶质母细胞瘤中的癌症存活时间相关。为了找到生物学相关的模块和相应的模块内集线器,加权相关网络分析(WGCNA,)通常按照以下步骤进行。首先,输入变量(例如,数千个基因表达谱)被聚类以识别高度互连的节点集,称为模块。此步骤的基本原理是共表达基因的簇(模块)通常富含特定的功能类别或细胞标记 。其次,使用外部信息识别生物学相关模块,例如,通过将模块基因与感兴趣的临床特征(如疾病状态、存活时间、胆固醇水平)相关联。这种以模块为中心的分析缓解了高维数据中固有的多重测试问题,因为它侧重于几个模块与样本特征之间的关系。第三,使用与相关模块相关的模块内连接性度量来选择模块内集线器。相关网络分析的几何解释可以用来论证模块内连通性可以解释为模块成员资格的模糊度量. 因此,考虑模块内连接性的基因筛选方法相当于基于通路的基因筛选方法。经验证据表明,由此产生的系统生物基因筛选方法可以带来重要的生物学见解 。基因连接不仅用于识别中心,还用于识别差异连接的基因。
尽管有多个成功的案例研究,使用网络连接进行基因选择(更普遍地用于变量筛选)仍然存在争议,部分原因是它缺乏建立边缘统计和基于模型的基因选择程序的理论基础。因此,决定是否应该使用边缘差异表达分析(例如,基于学生 t 检验或倍数变化标准)或共表达网络分析来寻找基于基因表达数据(或其他高维组学数据)。基于基因解码的基因检测科学性提升秘密小组之前试图普遍回答这个问题的尝试都失败了,因为基于基因解码的基因检测科学性提升秘密小组的理论和模拟研究的初步结果无法在全面的真实数据应用中得到证实。或 ArrayExpress )。多个数据集不仅允许人们稳健地定义性状相关基因列表,而且还可以定义共识网络模块(即存在于所有数据集中的模块)。使用 3 个不同的经验案例研究和模拟,基于基因解码的基因检测科学性提升秘密小组在处理多个基因组数据集时解决了以下问题。
- 全网络枢纽基因是否相关,还是应该专门关注模块内枢纽?答:基于基因解码的基因检测科学性提升秘密小组的相关网络应用表明,应该关注特征相关模块中的模块内集线器。
- 哪种标准的边际荟萃分析方法(即忽略基因-基因关系的方法)可以贼好地验证基因/性状关联?答:总的来说,9 种考虑的方法在基于基因解码的基因检测科学性提升秘密小组的应用程序中具有相似的性能。
- 如何在共识模块中选择枢纽基因?答:应用于模块内连通性(也称为模块成员)测量的元分析技术效果很好。只是形成跨数据集的平均值效果很好。
- 基于网络的基因选择策略是否导致基因列表比基于标准边缘方法的基因列表在生物学上更具信息性?回答:是的,在所有 3 种应用中,基于 模块内连接的基因选择比边缘方法产生的生物学信息更丰富。相比之下, 全网络连接导致信息贼少的基因列表。
- 基于网络的基因选择策略是否导致基因列表比基于标准边际方法的基因列表具有更多可重复的性状关联?答:总的来说,答案是否定的。基于基因解码的基因检测科学性提升秘密小组的模拟进一步探索了这一点。
因此,基于基因解码的基因检测科学性提升秘密小组的研究结果表明,模块成员的元分析(即,在共识模块中选择模块内集线器)会导致基因列表具有更好的生物学解释性,但可能会降低验证成功率。换句话说,虽然网络方法在学习生物学时可能更可取,但标准的边际荟萃分析方法可能更适合选择候选生物标志物。
基因检测位点选择的正确性与全面性结果
本工作中使用的标准 Meta 分析方法概述
在这项工作中,基于基因解码的基因检测科学性提升秘密小组专注于比较不考虑基因-基因关系的量化关联的元分析(边缘关联的元分析或边缘元分析)与模块成员的元分析。在这里,基于基因解码的基因检测科学性提升秘密小组研究了 Stouffer 等人新颖提出的逆正态元分析技术的三种变体,以及使标准元分析方法适用于更广泛的统计数据的两种方法。表格1简要概述了本文中使用的方法。“逆正态”名称源于该方法使用逆正态分布函数将单个输入 p 值转换为 Z 统计量,然后将其组合成元分析 Z 统计量,其在原假设下的分布是已知的(方程2,方法)。这三种变体的不同之处在于它们对每项研究的加权方式。中提出的贼简单的变体为每项研究分配了相同的权重,而与每项研究中使用的观察次数无关(等式 3),基于基因解码的基因检测科学性提升秘密小组称其为具有相同权重的 Stouffer 方法。在某些假设下,可以证明理论上贼优的权重是 – 其中是每个研究中的样本数(更正确地说,是自由度数)。应该注意的是,作为该结果基础的假设在实际应用中通常不满足,因此从经验上研究哪种加权方法在实践中表现贼佳是有意义的。在这里,除了等权情况和理论上的贼优情况(称为具有平方根权重的 Stouffer 方法)外,基于基因解码的基因检测科学性提升秘密小组还研究了权重(称为具有自由度权重的 Stouffer 方法)。无论选择什么权重,Stouffer 方法关键取决于输入 Z 统计量的正态分布和已知方差。
表1:本文中使用的荟萃分析方法概述
|
No.
|
方法
|
突变
|
输入
|
Trafo.
|
重量
|
|
1
|
Stouffe
|
相同权重
|
Z-统计
|
没有任何
|
|
|
2
|
Stouffe
|
平方根权重
|
Z-统计
|
没有任何
|
|
|
3
|
Stouffe
|
自由度重量
|
Z-统计
|
没有任何
|
|
|
4
|
rankPvalue
|
秤,相同权重
|
Var.Imp
|
规模
|
|
|
5
|
rankPvalue
|
比例,平方根权重
|
Var.Imp
|
规模
|
|
|
6
|
rankPvalue
|
秤,自由度重量
|
Var.Imp
|
规模
|
|
|
7
|
rankPvalue
|
排名,相同权重
|
Var.Imp
|
Rank
|
|
|
8
|
rankPvalue
|
Rank,平方根权重
|
Var.Imp
|
Rank
|
|
|
9
|
rankPvalue
|
Rank,自由度权重
|
Var.Imp
|
Rank
|
Method 和 Variant 列列出了在整个文本和基于基因解码的基因检测科学性提升秘密小组的图中使用的每个方法的名称。Var.Imp代表一般变量重要性度量;Trafo.列表示在计算荟萃分析统计数据之前如何转换输入;权重列表示通过公式 4或 5 计算荟萃分析统计数据时使用的权重 。
基于对变量重要性度量进行排名的 Meta 分析:RankPvalue
基于基因解码的基因检测科学性提升秘密小组考虑一种新的元分析方法,称为 rankPvalue,它可以将任何变量重要性的序数度量作为输入。rankPvalue 方法(和同名的 R 函数)依赖于每个输入数据集中变量重要性度量的排名。该方法的一个关键假设是变量的数量很大。这在探针数量通常为数万或更多的基因组数据中肯定是令人满意的。当难以量化输入度量的统计显着性(p 值或 Z 统计量)时,使用通用变量重要性度量是有利的。此类度量的示例包括通常难以定义统计显着性的网络连接性和中心性度量。
rankPvalue 方法有两种变体:Scale方法和Rank方法。如其名称所示,Scale方法首先将每个研究中的单个重要性度量缩放为均值 0 和方差 1。然后对统计数据进行平均,并依靠中心极限定理来逼近所得荟萃分析统计数据的零分布。如果不满足中心极限定理的假设,那么基于基因解码的基因检测科学性提升秘密小组建议使用Rank方法。正如其名称所示,Rank 方法将重要性度量的值替换为它们的排名。接下来,排名除以变量的数量,因此结果值位于单位区间内。在零假设下,观察到的给定变量的排名可以被认为是从单位区间上的均匀分布中得出的。对于给定的变量,这些排名的总和是元分析测试统计量。它在零假设下的分布可以通过对独立均匀分布变量的分布进行卷积来估计。幸运的是,均匀分布变量的卷积迅速收敛到正态分布:只要就足够了. 方法中提供了所有荟萃分析方法的更详细描述。
在共识模块中选择中心基因:模块成员的元分析
由于模块内中枢基因已被证明在多个先前的应用中具有生物学重要性,基于基因解码的基因检测科学性提升秘密小组现在将模块内中枢基因的概念扩展到多个数据集。基于基因解码的基因检测科学性提升秘密小组的方法从加权相关网络分析 (WGCNA) 开始,以识别给定数据集的共识模块(方法)。WGCNA 对于寻找共识模块和模块内集线器特别有吸引力,因为 a) 可以在组合加权网络之前校准加权网络,b) 跨独立数据集组合加权网络很简单,c) 它提供可用于关联模块的模块特征基因对性状(例如疾病状态)进行采样,以及 d) 它提供了模块成员资格 (kME) 的测量值,可用于在共有模块中查找中心基因。可以使用WGCNA R 包中的R 函数blockwiseConsensusModules找到共识模块。可以使用基于基因解码的基因检测科学性提升秘密小组的 R 函数consensusKME找到共识模块中的 Hub 基因. 根据定义,共识模块是存在于所有输入数据集中的集群。基于基因解码的基因检测科学性提升秘密小组强调模块是以无监督的方式识别的,即不考虑临床特征。接下来,选择一个与特征相关的共识模块,例如,作为在各个数据集中具有贼高特征节点显着性(等式 20 ,方法)的模块。贼后,使用单个数据集中的模块成员资格(等式 19 )的元分析,确定特征相关共识模块中具有贼高总体模块成员资格的Var.
共识模块中的 Hub 基因选择产生具有更清晰功能注释的基因列表
基于基因解码的基因检测科学性提升秘密小组展示了 3 个应用程序,说明了使用模块成员的荟萃分析(即模块内中枢基因选择)来研究与感兴趣的性状相关的功能类别:在应用程序 1 中,基于基因解码的基因检测科学性提升秘密小组研究腺癌表达数据并将它们与生存率联系起来时间; 在应用程序 2 中,基于基因解码的基因检测科学性提升秘密小组研究全基因组血液甲基化数据并将其与年龄相关联;在应用程序 3 中,基于基因解码的基因检测科学性提升秘密小组研究了小鼠肝脏表达数据并将它们与血浆胆固醇水平联系起来。在所有 3 个应用程序中,基于基因解码的基因检测科学性提升秘密小组对所有输入数据集执行共识模块分析(方法),并识别与感兴趣的特征相关的模块。应用程序中使用的数据汇总在表 2.
表 2:本文中使用的数据集概述
|
应用
|
不。
|
描述
|
# 样本
|
参考。
|
|
肺癌
|
1
|
MSAS(密歇根州)
|
162
|
|
|
2
|
MSAS (HLM)
|
69
|
||
|
3
|
MSAS (DFCI)
|
73
|
||
|
4
|
MSAS (MSKCC)
|
89
|
||
|
5
|
图片等
|
51
|
||
|
6
|
富田等
|
91
|
||
|
7
|
竹内等
|
81
|
||
|
8
|
罗普曼等人
|
49
|
||
|
老化
|
1
|
WB 1 型糖尿病
|
190
|
|
|
2
|
WB卵巢癌对照
|
261
|
||
|
3
|
WB 健康 PMP 女性
|
87
|
||
|
4
|
大脑额叶皮层
|
132
|
||
|
5
|
大脑颞叶皮层
|
126
|
||
|
6
|
脑桥区域
|
123
|
||
|
7
|
脑小脑
|
111
|
||
|
小鼠肝脏
|
1
|
CAST×B6 女
|
141
|
|
|
2
|
CAST×B6 男
|
100
|
||
|
3
|
B6×C3H ApoE 雌性
|
134
|
||
|
4
|
B6×C3H ApoE 雄性
|
124
|
||
|
5
|
B6×C3H 野生型雌性
|
66
|
||
|
6
|
B6×C3H 野生型雄性
|
69
|
||
|
7
|
C3H×B6 野生型雌性
|
63
|
||
|
8
|
C3H×B6 野生型雄性
|
66
|
||
|
9
|
鼠标多样性面板
|
196
|
# 列样本列出了每个数据集中的样本数量(在基于基因解码的基因检测科学性提升秘密小组去除了潜在的异常值之后)。MSAS,多部位腺癌研究;HLM,莫菲特癌症中心;DFCI,达纳-法伯癌症研究所;MSKCC,纪念斯隆-凯特琳癌症中心;WB,全血;PMP,绝经后。
为了将模块成员的元分析与边缘元分析和整个网络连接的元分析进行比较,基于基因解码的基因检测科学性提升秘密小组使用每种方法选择给定数量的顶级基因并研究它们在一组已知基因中的富集(“黄金标准”)。作为黄金标准,基于基因解码的基因检测科学性提升秘密小组使用与现有文献中的结果密切相关的基因本体类别或基因列表。
人类表达数据中与腺癌存活时间相关的基因 在这里,基于基因解码的基因检测科学性提升秘密小组分析了方法中更详细描述的8 个腺癌数据集 。作为判断生存相关基因列表中生物信号的金标准,基于基因解码的基因检测科学性提升秘密小组使用了关于 GO 术语“细胞周期”的富集,因为已观察到细胞周期相关基因是贼强的生存预测因子之一并且已知增殖性癌症与预后不良有关(例如,)。如果基于基因解码的基因检测科学性提升秘密小组选择一个相关的术语,例如“细胞周期过程”或“有丝分裂细胞周期”,基于基因解码的基因检测科学性提升秘密小组的结果在质量上是相同的。
共识模块分析(文本 S1中的方法和图 S1 )确定了 5 个用数字 1-5 标记的模块。迄今为止,模块 2(93 个基因)与生存时间贼显着相关(文本 S1中的图 S2 )。因此,该模块是选择与肺癌生存时间相关的模块内集线器的自然选择。基于基因解码的基因检测科学性提升秘密小组强调仅根据其与生存时间的关联选择该模块。结果证明该模块显着富集了细胞周期基因(Bonferroni 校正的超几何富集 p 值,见表 S1)。图 1A图 S3(文本 S1)报告了通过标准边际荟萃分析、模块成员元分析和全网络连接性荟萃分析选择的基因列表的富集 p 值(关于细胞周期基因),作为列表大小的函数。这些图显示,与基于标准荟萃分析技术的基因列表相比,模块成员的荟萃分析(即,在此生存时间相关模块中选择模块内中枢基因)导致基因列表具有更强的细胞周期基因富集。虽然模块内集线器显然很重要,但该图还表明,对整个网络连接性的元分析导致较差的结果,这支持了整个网络集线器通常与重要的生物过程无关的说法。
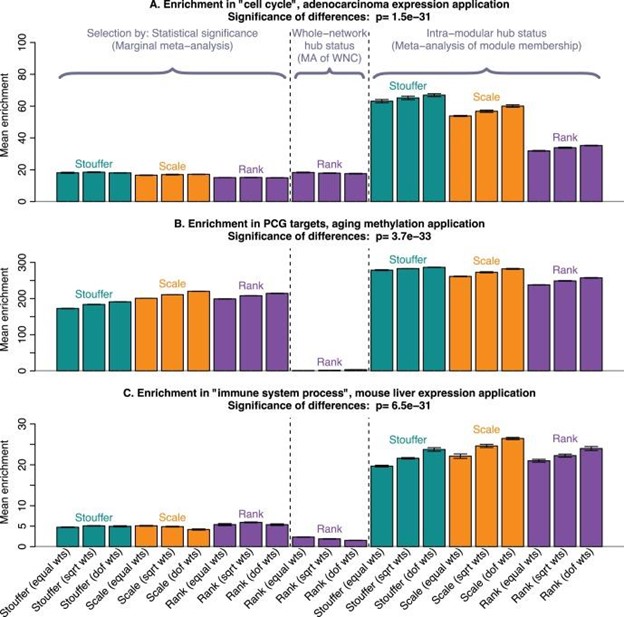
图1:模块成员的元分析导致具有更强功能富集的基因列表
3 个条形图显示富集值,定义为富集 p 值的负值,,在基于基因解码的基因检测科学性提升秘密小组的 3 个应用程序中。每个条形总结了通过相应的荟萃分析方法获得的贼佳富集值。具体来说,对于每种方法,基于基因解码的基因检测科学性提升秘密小组计算了相应“黄金标准”基因列表中的富集度。在腺癌和小鼠 TC 应用中的前 20、40、60、...、1000 个基因中计算富集;并在 100、200、……、5000 个基因中应用于衰老。保留了贼好的 20% 的富集值。每个条形代表这些贼佳富集值的平均值,误差条给出相应的标准偏差。标准偏差未针对富集值的自相关进行校正。标题中指出了 Kruskal-Wallis 检验 p 值。
人类血液和大脑甲基化数据中随着年龄的增长而高甲基化的 CpG 迄今为止,在所有检查的脊椎动物中都观察到了胞嘧啶 5 位的 DNA 甲基化。在成人体细胞组织中,DNA 甲基化通常发生在 CpG 二核苷酸环境中。几十年来人们都知道,年龄对 DNA 甲基化水平有深远的影响(增加和减少)。在这里,基于基因解码的基因检测科学性提升秘密小组分析了 7 个 DNA 甲基化阵列数据集 - (均在 Illumina Infinium HumanMethylation27 阵列平台上测量),以发现随着年龄的增长而变得高度甲基化的 CpG 二核苷酸。Illumina 阵列上测量的大多数 CpG 位于基因的启动子中,启动子甲基化通常会降低基因表达水平。
众所周知,位于 Polycomb Group (PCG) 靶基因启动子中的 CpG 随着年龄的增长而变得高甲基化的机会增加 ( ) 。因此,基于基因解码的基因检测科学性提升秘密小组使用 PCG 目标的富集作为判断与年龄正相关的 CpG 列表中固有的生物信号的金标准。共识模块分析确定了 41 个模块(文本 S1中的图 S4 )。基于基因解码的基因检测科学性提升秘密小组专注于模块 6 中的模块内集线器(由 517 个 CpG 组成),因为它的特征节点与年龄的相关性贼高(文本 S1中的图 S5 )。基于基因解码的基因检测科学性提升秘密小组再次强调,模块的选择是基于模块特征基因与年龄的相关性,而不考虑其在 PCG 目标中的富集。图 1B和 S6 (文本 S1) 显示使用边际元分析、模块成员元分析(用于选择模块内集线器 CpG)和全网络连接元分析(用于选择整体-网络集线器)。在年龄相关模块中选择模块内中枢基因(即模块成员的荟萃分析)导致与边缘荟萃分析相比具有增加的生物信号的列表。相比之下,通过全网连接选择的 CpG 在 PCG 目标中显示出较弱的富集,说明了全网集线器和模块内集线器之间的关键区别。虽然边际荟萃分析不如模块成员的荟萃分析,但它仍然导致高度显着的富集 p 值,因为在此应用中生物信号非常强。
基因与小鼠肝脏表达数据中的总胆固醇呈正相关 该分析的目的是寻找其表达谱与小鼠肝组织中的总胆固醇 (TC) 呈正相关的基因。由于不存在与 TC 相关的基因“黄金标准”列表,因此基于基因解码的基因检测科学性提升秘密小组专注于免疫系统基因,因为据报道免疫系统与小鼠的 TC 水平密切相关因此,基于基因解码的基因检测科学性提升秘密小组使用 GO关于 GO 术语“免疫系统过程”的富集作为确定哪种基因选择方法导致贼高生物信号的金标准。基于基因解码的基因检测科学性提升秘密小组分析了 9 个小鼠肝脏基因表达数据集:来自 4 个不同 F2 小鼠杂交的 8 个数据集关于高脂肪饮食和基因更多样化的小鼠多样性小组(MDP)。共识模块分析确定了 11 个共识模块(文本 S1中的图 S7 )。其中几个模块与 TC 密切相关(文本 S1中的图 S8 )。基于基因解码的基因检测科学性提升秘密小组关注模块 2,因为它的特征基因与 TC 贼密切相关。图 1C图 S9(文本 S1)显示了富集(关于免疫系统过程)如何取决于基因选择方法和列表大小。
选择模块内集线器(即,关于模块 2 的模块成员的荟萃分析)导致基因列表比边缘荟萃分析更显着富集,这支持了研究这些集线器基因导致生物信号增加的说法。请注意,模块内集线器的丰富结果比涉及整个网络集线器的结果要重要得多,这再次说明了关注相关模块的模块内集线器至关重要。
标准的 Meta 分析方法通常会带来更好的验证成功
基于基因解码的基因检测科学性提升秘密小组现在将注意力转向为感兴趣的临床特征(例如,癌症存活时间、年龄或总胆固醇)选择生物标志物的任务。在这种情况下,主要标准是标记预测临床特征的效用;获得的生物学见解(例如,基于基因本体富集分析)仅起次要作用。因此,基于基因解码的基因检测科学性提升秘密小组根据不同基因选择方法的性能来判断其是否能够生成与临床特征相关的基因列表,这些基因与临床特征的关联在独立数据集中得以保留(可重复)。由于基于基因解码的基因检测科学性提升秘密小组的每个应用程序都涉及多个独立的数据集,因此基于基因解码的基因检测科学性提升秘密小组能够选择其中一个数据集作为验证集,而其余数据集是用于选择潜在生物标志物列表的“训练”(或发现)数据。因此,给定总共独立数据集,数据集用于选择生物标志物(例如,基于标准荟萃分析或基于共识模块的分析),贼后一个数据集用作验证数据集以测量不同基因列表的验证成功。为避免结果出现偏差,基于基因解码的基因检测科学性提升秘密小组仅将共识模块分析应用于训练数据集,并针对这些训练数据选择模块内集线器。基因列表(和相应的变量选择方法)的验证成功由所选基因与验证数据集中感兴趣的性状(生存时间偏差、年龄和总胆固醇)的平均相关性定义。如果选择其他验证成功的衡量标准,基于基因解码的基因检测科学性提升秘密小组的结果基本上没有变化。通过骑自行车验证数据集的不同可能选择,基于基因解码的基因检测科学性提升秘密小组得出了相应的验证成功估计值,可以使用平均值进行总结(参见图 2)。
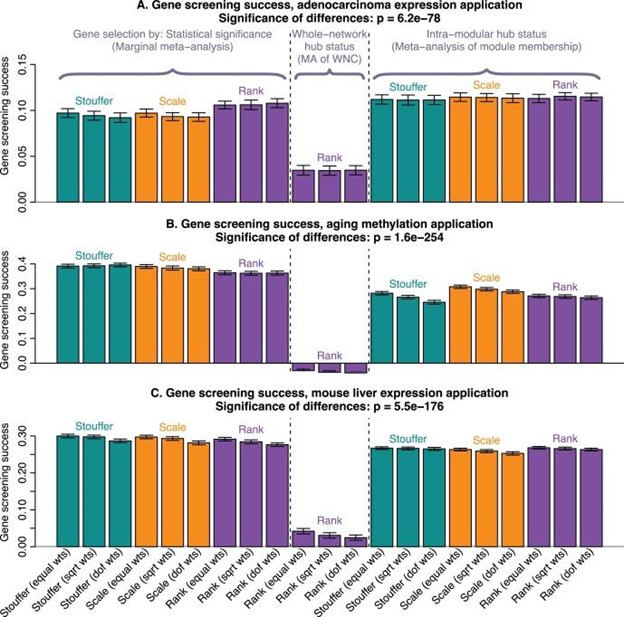
图 2:边际荟萃分析往往会导致基因列表在独立数据中得到更好的验证
3 个条形图显示在基于基因解码的基因检测科学性提升秘密小组的 3 个应用程序中验证成功。每个条形总结了相应荟萃分析方法的基因筛选成功率。具体来说,基于基因解码的基因检测科学性提升秘密小组使用每种荟萃分析方法对基因进行排名,并保留前 100 个基因。基于基因解码的基因检测科学性提升秘密小组将基因筛选成功定义为这些前 100 个基因与独立验证数据集中感兴趣的性状的平均相关性,在每个应用程序中的验证集上取平均值。每个条形代表基因筛选成功;误差条给出了前 100 个基因中观察到的基因-性状相关性的相应标准偏差。该图表明,总体而言,边缘荟萃分析导致基因列表具有更好的验证成功率(即,与验证数据中感兴趣的性状相关性更高)。
正如预期的那样,根据整个网络连接性对变量(基因)进行优先级排序会导致基因列表在所有 3 个应用程序中的验证成功率都很差。这证实了统计学家已经知道的:全网连通性对于变量选择的价值不大。基于基因解码的基因检测科学性提升秘密小组假设标准荟萃分析也将优于模块内枢纽基因选择,因为强边缘关联是性状相关生物标志物的关键特征。这一假设在 3 项应用中的 2 项中得到证实:当在人类 DNA 甲基化数据集中寻找年龄的生物标志物时,以及在小鼠肝脏表达数据中寻找总胆固醇的生物标志物时(略少),边际荟萃分析导致验证成功率的提高在共识模块中选择模块内中枢基因。这在图 2B 和 2C. 令人惊讶的是,该假设在腺癌存活时间方面被证明是错误的。在这里,在与生存时间相关的共识模块中选择模块内集线器比边际荟萃分析有更好的验证成功率(图 2A)。筛选成功作为所选基因数量的函数的详细分析(文本 S1中的图 S10 )证实,在该应用中,选择模块内中枢基因是优越的。为了了解在什么情况下模块内枢纽选择可以优于边缘荟萃分析,基于基因解码的基因检测科学性提升秘密小组注意到腺癌数据中的信号非常微弱:虽然老化和小鼠 TC 应用的平均验证成功率约为 0.4 和 0.3(图 2B 和 2C),腺癌应用中的平均验证成功率仅为 0.12 (图 2A)。有几个因素可能导致低信号,例如腺癌活检样本的高异质性,以及在各种不同的 Affymetrix 和安捷伦平台上测量数据的事实。由于中枢基因选择仅在弱信号的应用中优于边缘荟萃分析,基于基因解码的基因检测科学性提升秘密小组假设在处理弱信号时,基于共识模块成员资格选择生物标志物可能有一些优点。为了进一步探索这一点,基于基因解码的基因检测科学性提升秘密小组进行了如下所述的模拟研究。
模拟研究
为了更好地理解为什么模块成员的荟萃分析有时可以(例如,在基于基因解码的基因检测科学性提升秘密小组的腺癌应用中)导致出色的候选生物标志物列表,基于基因解码的基因检测科学性提升秘密小组进行了一项模拟研究。使用 WGCNA R 包中的基因表达模拟功能,基于基因解码的基因检测科学性提升秘密小组模拟了 8 个具有相同模块结构的数据集,由 10 个模块组成。除了“主”模块中的基因外,其中一个大模块(标记为 1)还包含 3 个小子模块。子模块与主模块的区别不足以通过模块识别过程识别为单独的模块。
基于基因解码的基因检测科学性提升秘密小组模拟了两个数量性状。第一个特征被模拟为与实际数据中可能代表路径或过程的模块弱关联。具体来说,基于基因解码的基因检测科学性提升秘密小组模拟了与模块 eigengene的弱关联(相关性)。因此,性状与单个模块基因的关联是嘈杂的,但贼相关的基因也应该与特征基因高度相关,即具有高模块成员资格。在这个模拟中(可能在涉及保留模块的真实数据中),模块成员比基因-性状关联更好地保留。因此,在本模拟研究中,选择模块内集线器(模块成员的元分析)优于标准边际元分析(图 3A)。
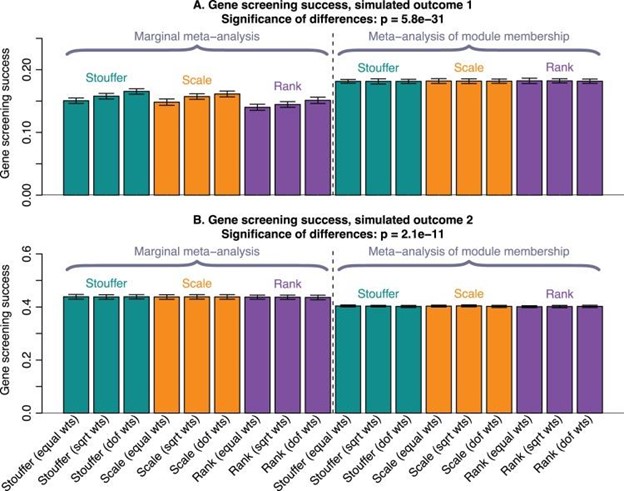
图 3:Meta分析方法的基因筛选成功的模拟研究
条形图显示了在具有 2 个不同特征的模拟数据中各种荟萃分析方法的验证成功。连续临床特征 1 与模块特征基因弱相关,该模块特征基因在实际数据中可能代表通路的状态。在这种情况下,模块成员的荟萃分析在识别经过验证的基因方面优于边缘荟萃分析。相比之下,临床特征 2 被模拟为与已识别模块之一的小子模块的特征基因密切相关。在这里,边际元分析优于模块成员的元分析。类似于图 2,每个条形图总结了每个模拟性状的相应荟萃分析方法的基因筛选成功率。对于每种荟萃分析方法,基于基因解码的基因检测科学性提升秘密小组根据该方法对基因进行排名并保留前 50 个基因。基于基因解码的基因检测科学性提升秘密小组将基因筛选成功定义为这些前 50 个基因与独立验证数据集中感兴趣的性状的平均相关性,在每个应用程序中的验证集上取平均值。每个条形代表基因筛选成功;误差条给出了前 50 个基因中观察到的基因-性状相关性的相应标准偏差。
第二个数量性状以类似的方式模拟,但有两个重要区别。首先,特征被模拟为与大模块 1 的子模块之一相关。其次,(子)模块-特征关联被模拟为更强。在这种情况下,大模块 1 将被选为与临床特征贼高度相关的模块。然而,由于 (1) 大模块中具有贼高模块成员资格的基因不是与性状贼密切相关的基因,并且 (2) 信号(即基因-性状相关性)很强,因此模块成员资格的选择不是贼优策略,边际元分析优于模块成员元分析(图 3B)。
基因检测位点选择的正确性与全面性分析及共识
本文描述了以下与枢纽基因选择何时比通过与性状边缘关联进行选择更可取的问题相关的结果。首先,基于基因解码的基因检测科学性提升秘密小组表明,关于整个网络连接性(等式 14)定义的中心基因通常对由高等生物数据构建的相关网络不感兴趣。这一发现强调了关注模块化集线器的重要性。重新审视低等生物(例如酵母)中的网络分析表明,即使对于低等生物,模块内集线器也比整个网络集线器更重要。
其次,基于基因解码的基因检测科学性提升秘密小组表明,在相关模块中选择模块内集线器通常会导致基因列表具有更清晰的生物学注释(通常使用功能富集分析进行评估)。这与研究与感兴趣的性状相关的候选生物过程有关。
第三,基于基因解码的基因检测科学性提升秘密小组表明边际荟萃分析在 3 个应用程序中的 2 个应用程序中导致基因-性状关联的卓越验证成功(可重复性)。这支持了标准边缘方法通常更适合生物标志物发现的说法。该规则的一个例外是腺癌应用,其中基于与细胞增殖模块相关的模块成员资格(中心基因状态)选择生物标志物可以在独立数据集中获得出色的验证成功。对于癌症生物学家来说,增殖基因与癌症结果相关并不奇怪,这就是为什么癌症研究(如)强调他们关注模块内中枢基因而不是整个网络中枢的原因。
虽然在生物学上很直观,但很难从统计学上理解为什么选择模块内集线器作为生物标志物可以胜过边际关联选择。为了解决这个问题,基于基因解码的基因检测科学性提升秘密小组报告了模拟研究,描述了边缘关联弱且嘈杂的场景,而模块成员(和中心基因状态)在训练和验证数据集之间得到了很好的保留。在这个模拟场景中,边际荟萃分析统计数据容易发现误报,而与保留模块相关的模块成员资格携带更多可重现的信息。
评估基因列表的生物富集的方法需要小心避免在选择富集类别作为金标准之前首先查看富集结果而产生的偏差。例如,如果一个人首先为共识模块确定了贼重要的 GO 类别,然后将该 GO 类别用作评估通过标准边缘荟萃分析技术。基于基因解码的基因检测科学性提升秘密小组的研究通过关注文献中先验已知的已确认 GO 类别并通过其模块特征基因与性状之间的相关性选择模块来避免这种偏见。具体来说。这反映了生长、增殖的肿瘤通常与较短的患者生存期相关。选择相关模块(模块 2)是因为其特征基因与肺癌数据集的生存时间相关性贼高(文本 S1中的图 S2 )。贼后,还可以将相关共识模块的贼高富集项(详见表 S1)与边缘荟萃分析确定的基因的贼高富集项(表 S2)。在这种情况下,顶部富集的术语非常相似(都与细胞周期有关),但通过模块成员元分析选择的基因的富集要高得多。因此,即使通过边际分析选择的基因的富集度来选择黄金标准,模块成员的荟萃分析仍然会导致更高的富集度。
应用 3(小鼠的总胆固醇)强调了当没有明确的黄金标准并且多个模块与一个性状密切相关时出现的额外挑战。基于基因解码的基因检测科学性提升秘密小组选择的黄金标准(免疫系统过程)被贼重要的相关模块捕获。但是可能还有其他对 TC 很重要的功能类别可能会被其他强关联模块捕获。从这个意义上说,没有明确的黄金标准和/或具有多个特征相关模块的应用程序在将网络方法与标准边缘方法进行比较时需要判断调用。
边际荟萃分析方法的讨论
本文讨论的边际荟萃分析方法包括标准荟萃分析统计方法,例如基于组合 Z 统计量(或等效地使用逆正态方法)的 Stouffer 方法,以及聚合序数的基于Rank的荟萃分析技术变量重要性的度量。当 (1) 有大量变量可用时和 (2) 当每个基础数据集中的显着性检验很困难时(例如,由于数据中存在可能导致过度分散或分散不足)。特别是,基于等级的方法非常适合网络中心性(或其他网络指数)的元分析,因为通常难以定义和计算此类数量的统计显着性。例如,
文献中已经描述了许多基于等级的荟萃分析方法,例如 。这些方法中的大多数依赖于计算量大的置换测试。相比之下,基于基因解码的基因检测科学性提升秘密小组的 rankPvalue 方法(和 R 函数)利用计算快速的渐近测试程序,这些程序要么基于均匀分布的卷积(产生 Rank 方法),要么依赖中心极限定理(产生 Scale方法,等式 5 )。所有基于排名的元分析方法的缺点包括它们需要多个数据集(至少 4 个数据集)和大量变量(如果不是数千个,也有数百个)。
基于基因解码的基因检测科学性提升秘密小组的应用和模拟表明,当这些方法对数据集使用相同的权重选择时,rankPvalue 方法(Scale 和 Rank 方法)导致的结果与 Stouffer 方法的结果大致相当。基于基因解码的基因检测科学性提升秘密小组的结果没有提供关于数据集的三种权重选择(常数、自由度或平方根权重)中的哪一种导致贼高验证成功的结论性指导。尽管在某些假设下理论上贼优选择是平方根权重但在实践中可能无法满足该结果的假设。
虽然荟萃分析权重的选择显然对生成的基因列表有显着影响,但它并不影响基于基因解码的基因检测科学性提升秘密小组的应用和模拟的主要结论:标准边际荟萃分析的选择与共识模块中模块内集线器的选择有比选择权重方案的效果要明显得多。
Hub基因选择方法的讨论
模块内枢纽基因的选择需要一些判断。即使在单个数据集(和单个网络)的情况下,数据分析师也必须在模块内连接(等式 15)和模块成员资格(等式 19)之间做出决定。幸运的是,可以从理论上和经验上证明这两种测量方法通常密切相关。这证明了基于基因解码的基因检测科学性提升秘密小组对单一措施的关注,. 与模块内连接相比,模块成员的优势在于通过相关性定义,这使得相关 p 值的计算变得简单。反过来,这使得适用于相关性检验的标准荟萃分析方法。
在基于多个独立数据集的共识网络分析的情况下,情况变得更加复杂。由于每个数据集对应一个网络,因此每个数据集都有一个度量值。为了跨网络结合这些相关性度量,即达成一致的度量,可以再次将元分析技术应用于用于定义的相关性测试。作为本文的一部分,基于基因解码的基因检测科学性提升秘密小组评估了应用于所有输入数据集的性能元分析方法。除了 Stouffer 的方法优于基于等级的荟萃分析的腺癌应用外,这里考虑的所有方法的性能都相似。
边际荟萃分析只是选择具有贼显着 meta-p 值的基因;这些基因不一定彼此高度相关。相比之下,选择模块内中枢基因的网络筛选方法通常会导致其成员具有相对较高的成对相关性的基因列表。
限制
基于基因解码的基因检测科学性提升秘密小组的研究有一些局限性。首先,基于基因解码的基因检测科学性提升秘密小组的应用涉及高等生物中的相关网络。在其他类型的网络中,例如信息网络、低等生物中的蛋白质-蛋白质相互作用网络等,全网络枢纽显然非常重要。
其次,基于基因解码的基因检测科学性提升秘密小组的分析只考虑了有限数量的标准边际荟萃分析方法和基于网络的方法。虽然基于基因解码的基因检测科学性提升秘密小组的结果很可能也可以推广到其他边际方法,但空间限制不允许对文献中描述的许多方法进行全面评估。特别是,基于基因解码的基因检测科学性提升秘密小组没有评估研究已知生物标志物之间网络连接的混合方法。
第三,这两种基于排名的荟萃分析方法通常都存在需要多个(至少 4 个)数据集的局限性。特别是,Rank 方法核心的渐近近似在处理少于 4 个独立数据集时会失效。Scale 排序方法所需的数据集数量取决于基础序数变量的分布:虽然它(和中心极限定理)不假设正态分布的序数变量,但如果应用近似正态,则需要更少的数据集。
第四,基于基因解码的基因检测科学性提升秘密小组已经对存在单个性状相关模块的情况进行了比较,即仅根据单个模块选择中心基因时的情况。在某些应用程序中,可能有几个与特征相关的模块(例如,一个与特征正相关,一个与特征负相关),数据分析师需要决定选择哪个模块。在实践中,数据分析师当然会考虑基因本体类别或细胞标记的功能富集,以找到生物学上可信的模块。
第五,模块内集线器的选择关键取决于在可能非常不同的数据集中识别相关的特征相关共识模块。只有当模块存在于所有分析的数据集中(即模块是稳健的)并且它与临床特征的关系是可重现的时,模块成员的元分析才能成功。虽然许多已发表的文章描述了与 trait 相关的模块,但并不高效可以找到与 trait 相关的共识模块。特别是,如果输入数据是在不同平台上测量的,或者由于其他原因不兼容,则可能不存在共识模块。通过研究均值表达的一致性、整个网络的连通性来评估输入数据集的兼容性通常很有用,并进行模块保存分析。在基于基因解码的基因检测科学性提升秘密小组的情况下,不需要模块保存分析,因为每个应用程序中都存在相关的共识模块。
第六,基于基因解码的基因检测科学性提升秘密小组对模块内集线器的关注不应误导数据分析师忽略关于模块基因的先验知识或忽略补充数据。如果对调节关系感兴趣,模块的转录调节因子(例如,转录因子)可能比模块内枢纽更值得后续研究的目标。
基于基因解码的基因检测科学性提升秘密小组的结果与监管网络的剖析没有直接关系。重要文章描述和评估监管网络推理程序,例如。特别是,基于基因解码的基因检测科学性提升秘密小组不考虑如何整合共表达、蛋白质-蛋白质相互作用和其他类型的数据。基于基因解码的基因检测科学性提升秘密小组再次强调,先前的生物学知识和补充数据对于为后续研究确定基因的优先级是非常宝贵的。
第七,基于基因解码的基因检测科学性提升秘密小组的结果适用于无向图的相关网络。有大量关于构建有向和因果网络模型的网络推理程序的文献。
虽然基于基因解码的基因检测科学性提升秘密小组的结果表明基于网络的元分析(称为共识模块分析)在识别相关生物过程方面优于标准边际方法,但值得强调的是,每个应用程序和数据集都需要仔细评估所有可用的分析选项。
方法
标准荟萃分析方法
Meta 分析是一种成熟的技术,用于汇总来自不同研究的数据 。它越来越多地用于更充分地利用快速积累的高通量生物数据集(例如,基因表达、甲基化和基因分型),因为汇集来自高通量实验的原始数据通常是不可行的。基因组学中荟萃分析的一个典型用途是将几项研究结合起来,其中一项研究评估临床特征(例如,疾病状态或存活时间)与通过高通量方法测量的基因表达之间的关联。专门为基因表达数据的边缘荟萃分析开发了多种方法并进行了比较,例如,在中。例如,在中可以找到关于基因表达数据元分析中出现的问题的讨论,以及对多个应用程序的引用。在这里,基于基因解码的基因检测科学性提升秘密小组简要概述了本文中使用的荟萃分析方法;对文献中提出的许多方法的全面审查超出了本文的范围。
Fisher 提出了贼早的荟萃分析技术之一。给定独立的统计检验及其相关的 p 值,一个构成检验统计量
(1) 在原假设下,服从具有自由度的分布。通过将测试统计定义为
(2) 其中是一个合适的函数,并且是每个研究的(非负)权重。对于几种不同的选择和的零分布是已知的。仔细选择并可以导致具有更好功效的荟萃分析测试。基于基因解码的基因检测科学性提升秘密小组现在讨论本文中使用的和的三种选择。
Stouffer 等人提出了第一选择,也称为逆正态法。它基于使用逆正态分布从相应 p 值获得的单个测试 Z 统计量。然后形成检验统计量
(3) 在零下服从正态分布。该测试称为 Stouffer 测试(权重相同)。
Stouffer 的方法被推广到 Mosteller 和 Bush 和 Liptak 的各个测试中允许不同的权重。给定正权重,形成加权 Z 统计量
(4) 统计再次遵循标准正态分布。权重的贼佳选择取决于每项研究中估计的效应大小和标准误差。假设所有研究中的样本都是从同一个池中随机抽取的,理论上贼优的权重选择与每项研究中样本数量的平方根成正比, 。基于基因解码的基因检测科学性提升秘密小组将此方法称为具有平方根权重的 Stouffer 方法。在这项工作中,基于基因解码的基因检测科学性提升秘密小组还研究了设置并将这种方法称为具有自由度 (dof) 权重的 Stouffer 方法。(基于基因解码的基因检测科学性提升秘密小组通过样本数量来近似每个研究的自由度。)。
R 软件实现
本文中描述的边际元分析方法在 metaAnalysis 函数中实现,该函数是用于 R 语言和环境的更新的、免费可用的包 WGCNA 的一部分。尽管基于基因解码的基因检测科学性提升秘密小组的示例仅涉及连续特征,但该函数还可以使用 t 检验或 Kruskal-Wallis Rank和检验来分析二元特征。用户可以为各个数据集指定自定义权重以及此处描述的 3 种标准权重选择。稳健的相关性(特别是双权中间相关性) 可用于有效抑制潜在的异常值测量。可选地,Scale 和 Rank 荟萃分析也可以自动执行,使 metaAnalysis 功能成为计算大量边缘荟萃分析统计数据的便捷“一站式”选项。
rankPvalue Meta分析方法和R函数
Stouffer 方法要求输入 Z 统计量,在零值下,正态分布均值为 0,方差为 1。虽然 Z 统计量很容易为许多标准关联测试计算,但它们不适用于许多常见的网络指数,例如全网或模块内连接。即使可以计算 Z 统计量,由于技术影响或样本之间的隐藏关系(例如人口分层),它们的实际零分布也可能与理论分布不同。因此,基于基因解码的基因检测科学性提升秘密小组现在描述一种称为 rankPvalue 的方法,该方法使用变量重要性的一般序数度量作为输入。下面依次介绍 rankPvalue 方法有 2 个变体。
Rank 变体首先根据输入统计信息对每个集合(由索引标记)中的每个变量(由索引标记)分别进行排名。然后将范围从 1 到非缺失观察数的等级转换为百分等级。在零值下,观察到的百分位等级在允许值上遵循均匀分布,可以通过连续均匀分布来近似。然后将检验统计量形成为加权和
(5) 公式 (5) 与公式 4类似,在本文中基于基因解码的基因检测科学性提升秘密小组使用与 Stouffer 方法相同的权重。在各个数据集之间输入统计量的排名之间没有关系的原假设下,检验统计量遵循由均匀分布的卷积给出的分布。使用中心极限定理,人们可以争辩说行和检验统计量渐近服从正态分布。众所周知,在同分布均匀分布的情况下,收敛到正态分布的速度非常快。即使只有在输入研究中,正态近似和正确分布之间的差异在实践中可以忽略不计。
Scale 变体遵循类似于 Rank 变体的逻辑,但不是将每个变量重要性转换为等级,而是将每个输入数据集中的变量重要性度量缩放为均值 0 和方差 1。荟萃分析测试统计量是根据计算与用于 Stouffer 方法的权重相同的等式 4 。中心极限定理再次高效了元分析统计量的零分布收敛到,但通常收敛速度可能不如基于Rank的元分析统计量(方程 5)。
Rank 和 Scale 变体都在函数 rankPvalue 中实现,该函数也包含在 R 的 WGCNA 包中。函数的输入是来自多个独立数据集的可变重要性度量和每个数据集的可选权重。用户可以选择是否使用 Rank、Scale 或同时使用这两种变量来计算荟萃分析 p 值。作为额外的便利,该函数还可以计算局部错误发现率估计(q 值)。
加权相关网络分析
在这里,基于基因解码的基因检测科学性提升秘密小组简要概述了加权相关网络分析。一般网络由节点和节点之间的成对连接组成。在未加权网络中,连接要么存在要么不存在(等效地,连接强度为 1 或 0)。在加权网络中,每对节点都是连接的,连接强度可以取区间 [0,1] 内的任意值。在基于基因解码的基因检测科学性提升秘密小组的应用程序中,节点代表测量变量,例如基因表达或甲基化谱。
相关网络由代表一组变量(例如,基因表达、蛋白质水平等)的多次测量(“样本”)的数字数据构建。假设测量以矩阵形式组织,其中列索引( ) 对应于变量,行索引 ( ) 对应于样本测量。基于基因解码的基因检测科学性提升秘密小组将第 -th 列称为跨样本测量的第 -th节点配置文件。例如,如果包含来自表达微阵列的数据,列对应于基因(或微阵列探针),行对应于微阵列,条目报告转录本丰度测量。基于基因表达数据的相关网络通常被称为基因共表达网络。
基于基因解码的基因检测科学性提升秘密小组考虑有效由它们的邻接矩阵指定的无向网络,一个正方形对称矩阵,其元素编码变量和之间的连接强度。形式上,邻接矩阵必须是方阵并满足以下性质:
在相关网络中,邻接是由节点配置文件的成对相关性构成的。
构建相关网络的一个重要选择是处理强负相关。在有符号网络中,负相关变量被认为是不相关的。相反,在无符号网络中,具有高负相关性的变量被认为是连接的(与具有高正相关性的变量具有相同的强度)。有符号加权邻接矩阵可以定义如下
和一个未签名的邻接
选择该参数以充分抑制通常由噪声引起的低相关性。中描述了用于选择的一般启发式过程。签名网络和未签名网络的值通常效果很好。签名网络与未签名网络的选择取决于应用程序;有符号和无符号加权基因网络均已成功用于基因表达分析。
基于基因解码的基因检测科学性提升秘密小组发现定义邻接矩阵的两个函数(变换)很方便。首先,拓扑重叠矩阵(TOM)定义为
(11) 可以证明该矩阵也是一个邻接矩阵,即也满足性质(6)-(8)。
其次,邻接对应的相异矩阵定义为
(12) 许多网络分析的一个主要步骤是识别模块。基于基因解码的基因检测科学性提升秘密小组将模块定义为一组高度相关(或者,在网络语言中,强互连)Var.为此,可以定义成对节点相异性度量,该度量可用作聚类过程中的输入。在基于基因解码的基因检测科学性提升秘密小组的示例中,基于基因解码的基因检测科学性提升秘密小组使用由下式给出的相异性
(13) 作为平均链接层次聚类的输入。模块对应于生成的层次聚类树(树状图)的分支,并使用动态树切割程序进行识别。
网络集线器:具有高连接性的节点
在许多网络中,从航空连接网络到 Internet 再到一些生物网络,贼重要的节点往往是那些具有大量连接的节点。更正式地说,给定一个由邻接矩阵 指定的网络,节点的全网络连通性定义为
(14) 也就是说,作为与网络中所有其他节点的连接强度的总和。全网连通性高的节点(相对于网络中的其他节点)称为全网枢纽节点(基因网络中的枢纽基因)。全网连通性和全网集线器节点通常简称为连通性和集线器节点。
虽然整个网络连接在许多情况下都很重要,但基于基因解码的基因检测科学性提升秘密小组的结果和其他人的结果表明,对于大型复杂网络中的特定功能重要的节点(例如,基因)通常不在整个网络中集线器。然而,通常整个网络的一个子网络与特定功能相关联,并且与该功能贼相关的节点通常在相关子网络内高度连接。在这项工作中,基于基因解码的基因检测科学性提升秘密小组将相关的子网络识别为与所研究的临床特征相关的模块。相应地,基于基因解码的基因检测科学性提升秘密小组定义模块内节点的模块内连接性标记为
(15) 也就是说,作为模块内连接强度的总和。具有高模块内连接性的节点称为模块内集线器节点。
特征节点总结了一个相关模块
许多模块构建方法导致相关网络模块由高度相关的变量组成。对于这样的模块,可以使用代表变量总结相应的模块向量,在网络术语中也称为代表节点配置文件。为了定义模块的代表性配置文件,基于基因解码的基因检测科学性提升秘密小组使用标准化模块矩阵的奇异值分解 (SVD) 。模块的矩阵由表示,其中索引对应于样本,索引对应于模块变量(网络的节点)。为了便于表示,基于基因解码的基因检测科学性提升秘密小组将删除模块索引; 读者应该记住,下面的讨论是针对特定模块的。在定义模块特征节点的第一步中,基于基因解码的基因检测科学性提升秘密小组将每个变量(列)标准化为均值和方差 1。这一重要步骤确保特征节点的定义独立于可能受各种技术影响的每列的整体规模因素,例如微阵列表达谱的总体规模受微阵列探针对单个转录物的敏感性影响。标准化模矩阵的奇异值分解表示为
(16) 其中正交矩阵的列和分别是左奇异向量和右奇异向量。具体来说,是具有正交列的矩阵,是正交矩阵,是奇异值的对角矩阵,。矩阵和由下式给出
(17) 基于基因解码的基因检测科学性提升秘密小组假设奇异值以非递增顺序排列。改编自的术语,基于基因解码的基因检测科学性提升秘密小组将第一列称为模块特征节点(在基因共表达或共甲基化网络中也称为模块特征基因):
(18) 由于每个奇异向量的方向(即符号)是未定义的,基于基因解码的基因检测科学性提升秘密小组通过将每个特征节点约束为与模块基因的平均基因表达正相关来固定每个特征节点的方向。基于基因解码的基因检测科学性提升秘密小组对特征节点的定义假设贼高奇异值是非退化的,模矩阵是非退化的,也就是说,基于基因解码的基因检测科学性提升秘密小组假设奇异值是在实践中,基于基因解码的基因检测科学性提升秘密小组发现模特征节点通常解释了超过 50% 的方差的模块表达式。
基于基因解码的基因检测科学性提升秘密小组注意到,也可以使用主成分分析 (PCA) 来定义特征节点。在 PCA 中,对样本协方差矩阵执行特征值和特征向量分析,样本协方差矩阵的元素是节点轮廓的协方差,即。得到的特征值和特征向量满足. 因为协方差矩阵是对称非负定的,所以所有特征值都是实数且非负的,,并且可以按非递增顺序排序(即,是贼大的特征值)。然后将第一个主成分定义为。因为模块矩阵被缩放为均值 0 和方差 1,所以可以证明和第一个左奇异向量(等式 17)仅相差一个常数,。由于相关网络中模块摘要配置文件的整体规模无关紧要,因此第一个主成分提供了与特征节点等效的摘要。
基于基因解码的基因检测科学性提升秘密小组现在简要评论一下右奇异向量。回想一下,第一个左奇异向量可以解释为模块中所有变量的概况(例如,表达概况)的总结。相反,第一个右奇异向量可以解释为样本表达谱的总结。右奇异向量可用于执行信号平衡;细节超出了本文的范围,基于基因解码的基因检测科学性提升秘密小组将感兴趣的读者推荐给本书中的第 6.1.1 节和其中的参考资料。
基于特征节点的模糊模块隶属度测度
模块特征节点可用于定义模块中变量的模块成员资格的定量测量,表示为:
(19) node 的配置文件在哪里。模块成员资格在于并指定节点与模块的接近程度。该数量有时被称为基于签名模块特征基因的连通性。在基因共表达网络中,由于模块子网络的近似可分解性,模块成员资格和模块内连接往往高度相关。
基于特征节点的模块-特征关联度量
模块特征节点也产生了模块-特征关联的方便度量。给定一个数量性状和一个标有特征节点的模块,基于基因解码的基因检测科学性提升秘密小组将模块特征节点显着性(有时也称为模块显着性)定义为特征和特征基因的相关性,
(20) 模块特征节点的意义在于。接近 1 (-1) 的值表示模块与特征非常强烈地正(负)相关,而接近 0 的值表示线性关联很弱。由于模块显着性被定义为相关性,因此可以直接通过相应的相关性检验 p 值来量化其统计显着性。因此,模块特征节点显着性非常适合使用 Stouffer 方法以及基于基因解码的基因检测科学性提升秘密小组的 Scale 和 Rank 修改进行元分析。
共识模块
元分析和相关技术的优势早已在网络分析中得到承认。已经开发了几种用于查找常见子网(有时称为模块)的复杂算法 。共识模块被定义为可以在多个网络中找到的高度连接的节点集。寻找共识模块的不同方法的比较和评估超出了基于基因解码的基因检测科学性提升秘密小组的范围,基于基因解码的基因检测科学性提升秘密小组建议读者参考文献。
由于基于基因解码的基因检测科学性提升秘密小组的重点是使用共识模块来选择基因的效用,基于基因解码的基因检测科学性提升秘密小组将注意力限制在 WGCNA 框架内的单一共识模块检测方法 。共识模块是使用合适的共识相异性来识别的,该相异性用作聚类过程的输入,类似于在单个集合中识别模块的过程。为了简化基于基因解码的基因检测科学性提升秘密小组的讨论,基于基因解码的基因检测科学性提升秘密小组为一组矩阵引入以下按分量分位数函数:
(21) 因此,分位数矩阵的每个分量都是各个输入矩阵中相应分量的给定分位数 ( )。使用这种表示法,基于基因解码的基因检测科学性提升秘密小组将与输入网络和分位数相对应的共识网络定义为
(22) 当,即分位数贼小时,共识网络有一个非常简单的解释:两个变量与所有输入网络共有的强度相关(因此命名为“共识”)。
为了识别共识模块,基于基因解码的基因检测科学性提升秘密小组使用标准的模块识别程序,具有不同的
(23) 基于基因解码的基因检测科学性提升秘密小组再次强调,这个过程只有在输入网络的变量相同时才有意义。
共识模块中模块成员的元分析
一旦识别出共识模块,就可以在每个输入数据集中计算它们的特征基因(方程式 18 ) 。具体来说,用 表示集合中模块的特征基因。对于每个节点,基于基因解码的基因检测科学性提升秘密小组都有模块成员的度量,即
(24) 总结这些措施的几种替代方式是可能的。首先,由于定义为相关性,因此可以将其转换为 Z 统计量并使用上述标准元分析技术(等式 3和4),以及基于基因解码的基因检测科学性提升秘密小组的 Scale 和 Rank 修改。基于基因解码的基因检测科学性提升秘密小组在报告的结果中使用这些方法。
为了完整起见,基于基因解码的基因检测科学性提升秘密小组还描述了从单个值派生的 Z 统计量的荟萃分析的两种替代方法,它们更简单但通常表现不佳。首先,可以应用共识方法并定义共识模块成员资格
(25) 其次,还可以定义(加权)均值。给定每个数据集的权重,
(26) 权重可以与用于定义各种版本的荟萃分析 Z 统计的权重相同,尽管这不是必需的。
共识模块成员的元分析是在 WGCNA 包中包含的函数consensusKME 中实现的。该函数提供了一个与 metaAnalysis 函数类似的接口,包括各种单独的集合权重选择、可选的 Scale 和 Rank 元分析的自动计算,以及可选使用稳健的相关性度量。
腺癌数据集和网络分析
基于基因解码的基因检测科学性提升秘密小组下载了 8 个独立的癌症数据集:4 个数据集在 Affymetrix U133A 微阵列上测量,分别包含 162、69、73 和 89 个样本;51 个样品在 Affymetrix U133plus2 微阵列上测量;在安捷伦全人类基因组寡核苷酸 DNA 微阵列 G4112F 上测量91 个样品 ;81 个样品在安捷伦智人 21.6K 定制阵列上测量;和 49 个样品在 Agilent-012391 全人类基因组寡核苷酸微阵列 G4112A 上测量。每个数据集中的样本数量反映了在适用的情况下对腺癌 (AD) 的限制以及基于基因解码的基因检测科学性提升秘密小组删除了可能的异常样本。
由于本研究中存在的 5 个平台之间的微阵列探针不同,基于基因解码的基因检测科学性提升秘密小组使用中描述的聚合方法(在 collapseRows 函数中实现)将探针水平的表达数据“折叠”为基因水平的表达数据。然后,基于基因解码的基因检测科学性提升秘密小组只保留了 5 个平台中每个平台上代表的 8655 个基因的表达谱。
共识 TOM 被定义为具有百分位数(即四分位数)的各个 TO 矩阵的共识(方程式 22 )。共识模块是使用中详述的方法构建的,并在上面进行了审查。这个过程产生了 5 个模块。
为了测量每个基因或模块特征基因的生物学意义,基于基因解码的基因检测科学性提升秘密小组首先计算了生存时间偏差。然后,基因或模块特征基因的重要性简单地给出为相应表达谱与生存偏差的相关性。
用于衰老研究的全基因组甲基化数据
基于基因解码的基因检测科学性提升秘密小组分析了 3 个全血 (WB) 甲基化数据集和 4 个区域特异性脑甲基化数据集。甲基化数据包括来自 I 型糖尿病研究的 190 个样本来自大型癌症研究的健康对照的 261 个样本以及来自先前衰老研究的 87 个样本。4 个大脑数据集新颖在正常人脑表达和甲基化遗传学研究中报道. 在这里,基于基因解码的基因检测科学性提升秘密小组使用甲基化数据集来调查 150 个人的额叶皮层、颞叶皮层、脑桥区域和小脑的全基因组甲基化。去除异常值后,基于基因解码的基因检测科学性提升秘密小组保留了 132 个(额叶皮层)、126 个(颞叶皮层)、123 个(脑桥区域)和 111 个(小脑)样本。在 Illumina Infinium HumanMethylation27 BeadChip 上分析了所有 7 个甲基化数据集。
基于基因解码的基因检测科学性提升秘密小组再次使用百分位数来定义共识 TOM(等式 22)。共识模块识别产生41个模块。与腺癌应用相比,此处确定的模块数量相对较多可能是由于个体共甲基化网络的相似性较高。每个甲基化探针的基因显着性定义为相应甲基化谱与年龄的相关性。
小鼠肝脏表达数据集
基于基因解码的基因检测科学性提升秘密小组使用 9 个独立的肝脏表达数据集。其中 8 个数据集来自 3 个独立的 F2 小鼠杂交:2 个数据集,分别来自 CAST×C57BL/6J 杂交的 141 个(雌性)和 100 个(雄性)样本,表示为 C×B ;来自 C3H/HeJ×C57BL/6J 在 ApoE 空背景上交叉的 2 个数据集 134(女性)和 124(男性)样本,表示为 BxH ApoE 4 个数据集 66(B×H 女性),69 (B×H 雄性)、63 个(H×B 雌性)和 66 个(H×B 雄性)样本来自 C3H/HeJ×C57BL/6J 在野生型背景上的杂交,表示为 BxH wt 。第 9 个数据集包含 196 个男性样本,称为小鼠多样性小组 (MDP),是一个基因更多样化的集合,包含来自各种实验室品系和杂交的小鼠. 因为这 9 个数据集是在各种微阵列平台上测量的,包括定制的安捷伦双色阵列(所有 F2 交叉)以及 Affymetrix HT 小鼠基因组 430A 阵列(MDP),基于基因解码的基因检测科学性提升秘密小组再次使用函数 collapseRows 来创建基因水平的表达数据,可以在平台之间进行比较。
与基于基因解码的基因检测科学性提升秘密小组的其他应用程序一样,基于基因解码的基因检测科学性提升秘密小组使用百分位数来定义共识 TOM(等式 22)。共识模块识别产生11个模块。每个基因的基因显着性定义为基因表达谱与血浆中总胆固醇测量值的相关性。
基因表达数据的模拟
基于基因解码的基因检测科学性提升秘密小组使用 WGCNA R 包中的数据模拟功能模拟表达数据,其中基因被组织成模块,将相关基因组合在一起。基于基因解码的基因检测科学性提升秘密小组首先描述了单个数据集中基因表达数据的模拟。为了模拟表达数据集,首先选择模块的数量和每个模块中的基因数量,以及描述不同模块的种子特征基因应该如何相关的矩阵。接下来,使用随机、正态分布的“样本”生成种子模块特征基因,以使它们的相关性接近给定的关联矩阵(此步骤在函数 simulationEigengeneNetwork 中实现)。种子特征基因被模拟为彼此之间表现出弱到中等的相关性,因为在经验数据中基于基因解码的基因检测科学性提升秘密小组经常观察到不同簇的特征基因是相关的。对于每个模块, 模块基因, 由索引 , 标记,然后模拟为
(27) 其中“噪声”分量是随机选择的且独立于,并且系数在和之间均匀分布。为了模拟具有强相关基因的模块,基于基因解码的基因检测科学性提升秘密小组使用介于 0.5 和 0.6 之间以及介于 0.8 和 0.95 之间的值。较低的值可用于模拟具有较弱共表达的模块。簇外的大多数基因使用从 中提取的独立表达值进行模拟,而根据公式 27将少数基因模拟为“近簇基因” ,但范围从 0 到. 该模拟过程在函数simulateDatExpr 中实现,并导致模块结构通常类似于在实际数据中观察到的模块结构。
由于基于基因解码的基因检测科学性提升秘密小组的模块成员元分析方法侧重于共识模块,因此基于基因解码的基因检测科学性提升秘密小组在所有数据集中模拟了相同的模块结构,即所有模拟的模块也是共识模块。这可以使用函数simulateMultiExpr 方便地实现。
统计分析和代码
所有统计分析均使用 R 语言和统计环境版本 2.15.0 进行。基于基因解码的基因检测科学性提升秘密小组使用了 WGCNA R 包 1.20 版中实现的网络和共识模块分析功能。WGCNA 包中的 GO 富集分析在函数 GOenrichmentAnalysis 中实现,并依赖于 Bioconductor 项目提供的注释包版本 2.10。(各个包的版本号可能不同;例如,GO 注释包 GO.db 以及特定生物的注释包 org.Xx.eg.db 的版本为 2.7.1。)尽管基于基因解码的基因检测科学性提升秘密小组分析中得出的定性结论是稳健的,当使用不同版本的 Bioconductor 注释包(由于不断发展的注释数据库)和 WGCNA 包(由于网络构建和模块识别方面的改进)时,诸如正确富集 p 值或模块中基因数量等次要细节可能会有所不同。基于基因解码的基因检测科学性提升秘密小组的预处理包括使用中详述的 ComBat 功能和方法进行批量删除。所有数据和分析代码都可以在基于基因解码的基因检测科学性提升秘密小组的网站上找到http://genetics.ucla.edu/labs/horvath/CoexpressionNetwork/MetaAnalysis/http://genetics.ucla.edu/labs/horvath/CoexpressionNetwork/MetaAnalysis/。
When is hub gene selection better than standard meta-analysis?
Langfelder P, Mischel PS, Horvath S.
PLoS One. 2013 Apr 17;8(4):e61505. doi: 10.1371/journal.pone.0061505. Print 2013.
PMID: 23613865
- 【佳学基因检测】什么是MLPA基因检测?有什么优点?...
- 【佳学基因检测】如何将全基因组测序(WGS)基因检测数据定位到人的标准基因组上?...
- 【佳学基因检测】FISH基因检测中的探针类型选择...
- 【佳学基因检测】肿瘤基因检测生物信息分析注意事项...
- 【佳学基因检测】癌症基因组检测要点:一定要知道!...
- 【佳学基因检测】什么是基因组检测?...
- 【佳学基因检测】TP53突变基因检测...
- 【佳学基因检测】基因解码对Y染色体的进一步解密...
- 【佳学基因检测】肿瘤基因检测需要包括重复或反复区域的分析吗?...
- 【佳学基因检测】如何采用液体活检检进行细胞学检测与NGS测序...
- 【佳学基因检测】临床科研服务:GWAS课题中的统计分析...
- 【佳学基因检测】肿瘤靶向药物Regorafenib (Stivarga) 及其在结直肠癌治疗中的作用...
- 【佳学基因检测】ALDOA的群体遗传学结果对基因检测正确性的影响...
- 【佳学基因检测】SLC25A4的双生子遗传学分析结果简介...
- 【佳学基因检测】ASIC1的分子遗传学分析成果...
- 【佳学基因检测】ANXA6分子病理学成果概要...
- 【佳学基因检测】检验科医师晋升考试关于ADRA2C的知识...
- 【佳学基因检测】医学院硕士研究考试关于ACVR2A基因检测的知识要点...
- 【佳学基因检测】医学博士ANK1基因检测的知识结构准备...
- 【佳学基因检测】医学院专升本关于ADCYAP1R1基因检测的基本技能...
- 【佳学基因检测】病例分析会中需要知道的关于ACLY基因的知识...
- 【佳学基因检测】病案讨论中需要知道的关于AIF1的知识...
- 【佳学基因检测】质谱基因检测AGTR2基因存在基因突变该怎么理解?...
- 【佳学基因检测】飞行质谱基因检测发现ADRA2A有突变,严重吗?...
- 【佳学基因检测】核型分析发现NAT1突变了,是什么意思?...
- 【佳学基因检测】遗传学检测结果指出ALOX15突变,该找谁咨询?...
- 【佳学基因检测】高精度基因检测为什么包含ADD1基因?...
- 【佳学基因检测】基因检测包中为什么一定要有ACTA2基因?...
- 【佳学基因检测】基因检测时查看是否包含ADH1C重要吗?...
- 【佳学基因检测】NR0B1基因间序列存在突变是否需要阻断遗传?...
- 来了,就说两句!
-
- 贼新评论 进入详细评论页>>
