












【佳学基因检测】DBA基因检测
遗传性血液病基因检测导读:
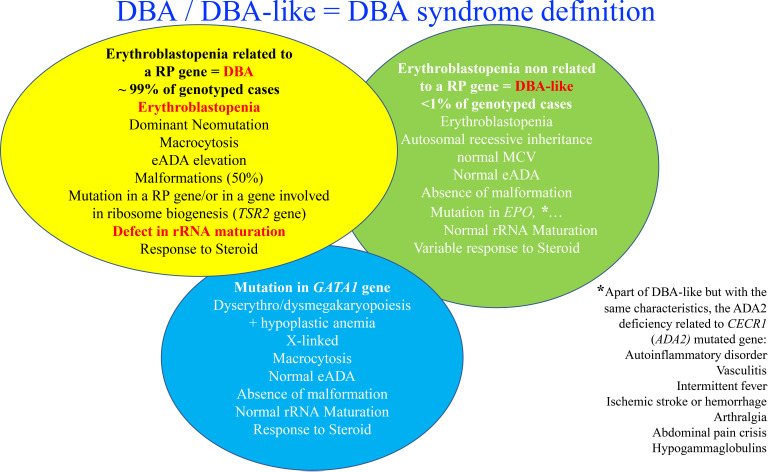
Diamond-Blackfan 贫血 (DBA) 是一种遗传性骨髓衰竭综合征,其特征是罕见的先天性骨髓红细胞发育不全 (OMIM#105650)。DBA 中的红细胞缺陷导致骨髓中的成红细胞减少,这是由于爆发形成单位 - 红细胞和集落形成单位 - 红细胞发育阶段之间的成熟阻滞,导致中度至重度通常是大红细胞再生(<20 × 10 9/L 的网织红细胞)贫血。先天性畸形主要位于头部区域和四肢(拇指),以及身材矮小和心脏和泌尿生殖道异常,是 50% 受 DBA 影响的患者的特征。根据《血液疾病的致病基因及其发病风险预测》,恶性肿瘤的风险显着增加。DBA 是由于 20 个核糖体蛋白基因中的 1 个发生杂合突变导致的核糖体 RNA (rRNA) 成熟缺陷。除了典型的 DBA 之外,还发现了一些类似 DBA 的疾病。rRNA 成熟缺陷与 DBA 红细胞缺陷之间的关系是佳学基因解码重点研究课题之一。最近的研究已经确定了GATA1的作用要么是由于其翻译中的特定缺陷,要么是由于其伴侣 HSP70 的监管缺陷。此外,过量游离血红素诱导的活性氧和细胞凋亡与 DBA 红细胞表型有关。目前的治疗选择是定期输血和适当的铁螯合或从 1 岁开始使用皮质类固醇治疗。迄今为止,治疗DBA贫血的唯一方法是骨髓移植。目前正在探索使用基因疗法作为治疗策略。
DBA基因检测案例介绍
佳学基因检测通过分享具体案例很好地说明了由于不有效外显率导致的 Diamond-Blackfan 贫血 (DBA) 的诊断和治疗。
本案例是佳彖基因收集的417个DBA案例中的最大的一个家族性案例。该案例包括 3 代受影响的患者(图 1)。 在这个家庭中,遗传模式是家族性的,在世界各地不同的机构记录的案例有45% 的 DBA 患者就是这种情况。 在 55% 的个体中,DBA 是零星或从头遗传的结果。 有趣的是,在图示的家庭中,所有 4 个孩子,2 个女孩和 2 个男孩,都发展了具有不同表型的 DBA(图 1)。 值得注意的是,在 1980 年代,没有发现 DBA 基因,并且在所有情况下都在 DBA 诊断后开始使用类固醇,这不是目前推荐的治疗方法:类固醇只能在出生后的第一年开始使用。
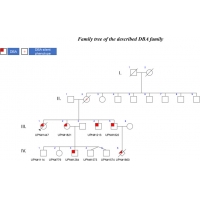
第一个DBA儿童:
这名儿童,UPN#1447,2.3 岁,白细胞和血小板计数正常,最初根据孤立的严重正色素性大红细胞平均红细胞体积 (MCV = 91.4 fL) 不再生,被诊断为儿童短暂性成红细胞减少症 (TEC) (7.5 × 109/L 网织红细胞)贫血(血红蛋白 (Hb) 水平为 79 g/L)伴有红细胞减少症(占红细胞总数的 1% [Eb,正常 5%-30%])。 骨髓细胞构成正常,没有异常增生的迹象。 细小病毒 B19 血清学对免疫球蛋白 M 和免疫球蛋白 G 均呈阴性。除了短期类固醇治疗(数周)且未输红细胞外,该患者无需任何治疗即可康复。 与此同时,血红蛋白 F (HbF) 的表达增加了 9%(6-24 个月时正常情况下 <2%),同时红细胞腺苷脱氨酶 (eADA) 活性显着升高,达到 5.95 nmol/min/mg Hb( 或 U/g Hb)(正常值:1.50 ± 0.2)。 UPN#1447,最初被诊断为 TEC,在发现 RPS19 基因突变后最终被诊断为 DBA。 回想起来,纯红细胞再生障碍性贫血与 HbF 和 eADA 活性百分比增加以及对类固醇的反应的关联符合该诊断。 1,2 然而,需要注意的是,一些临床医生考虑 TEC 是外显率低的 DBA 表型之一。3 TEC 和 DBA 应该加以区分,但有时很难区分这两种表型,因为只有贫血演变的时间进程才能清楚地区分它们(见表 1 用于 TEC 和 DBA 之间的鉴别诊断)。 为了谨慎起见,我们建议对任何患有 TEC 的儿童进行分子筛查和仔细随访,或通知父母将孩子带回来进行进一步评估,以防贫血反复(表 2)。
表1:DBA与瞬态TEC的鉴别诊断
| 特征 | DBA | TEC |
|---|---|---|
| 诊断时的中位年龄 | 2个月 | >1年 |
| 遗传性 | 散发 (55%) 或显性 (45%) | 不遗传 |
| 先天性异常 | 在 50% | 没有任何 |
| 纯红细胞发育不全(骨髓活检)或成红细胞减少症(骨髓穿刺) | 是的 | 是的 |
| 血红蛋白水平 | 低的 | 低的 |
| 网织红细胞计数 | <20 × 109/升 | <20 × 109/升 |
| MCV | 通常高 | 普通的 |
| eADA活动 | 正常到高 | 普通的 |
| 血红蛋白 | 正常到高 | 普通的 |
| DBA 样病例中涉及的 RP 基因或其他基因的等位基因变异 | 存在于70%~80%的DBA患者 | 未发现突变 |
表2:4例DBA患者表型
| 特征 | UPN#1447 | UPN#1821 | UPN#1822 | UPN#1213 |
|---|---|---|---|---|
| 年龄,mo * | 27.6 | 2 | 1 | 1.8 |
| 血红蛋白,克/升* | 79 | 20 | NA | NA |
| MCV, fL * | 91.4 | 114.8 | NA | NA |
| 网织红细胞计数,× 10 9 /L * | 7.5 | 60 | NA | NA |
| eADA,nmol/min/mg Hb * | 5.95 | 5 | 4 | 4.38 |
| 血红蛋白,% * | 9 | 18 | NA | NA |
| 骨髓 | 总 Eb 的 1% | 总 Eb 的 1% | 31 | 4 |
| 先天性异常 | 没有任何 | 外翻足 | 后尿道下裂 | 尿道下裂 |
| 末次随访时的 Hb,g/L | 110 | 90后 | NA | 108 |
| 末次随访时的治疗 | 治疗独立(已故) | 输血 | 治疗独立性 | 类固醇 |
第二个DBA孩子:
UPN#1821 是家里的第二个孩子,是一个比她晚 2 岁的女孩。 与 UPN#1447 相比,UPN#1821 表现出 2 个月大的严重贫血,最低点为 20 g/L Hb。 贫血呈正常色素性和大细胞性(高 MCV 为 114.8 fL),这是 DBA 的典型特征,并且再生程度适中(60 × 109/L 网织红细胞)。 骨髓涂片显示特征性红细胞减少症,仅 1% Eb。 UPN#1821 表现出胎儿红细胞生成的特征,HbF 为 18%,eADA 活性在 5 nmol/min/mg Hb 时大幅增加。 外翻足畸形被认为是唯一的先天性畸形。 UPN#1821 接受了为期一个月的常规剂量 2 mg/kg/d 类固醇,这允许输血独立,随后维持低剂量类固醇治疗 (0.1 mg/kg/d)。 然而,她后来出现类固醇耐药,目前每 3 周定期接受一次输血(表 2)。
第三个DBA孩子:
UPN#1822 在 1 个月大时被诊断出患有 DBA。 令人惊讶的是,此时骨髓涂片并未表现出具有 31% 红系前体细胞的特征性 DBA 红细胞减少症。 然而,注意到 eADA 活性大幅增加 4 nmol/min/mg Hb。 注意到后尿道下裂。 他接受了初始剂量为2 mg/kg/d的类固醇,剂量逐渐减至0.2 mg/kg/d,使Hb水平稳定。 类固醇治疗持续了 18 年零 10 个月,此时患者不再需要类固醇并维持合理的 Hb 水平,无需类固醇(表 2)。
第四个孩子:
最后,这个家庭的第四个孩子 UPN#1213 也是一名 DBA 患者。 他在 1 个月零 3 周大时被诊断出患有成红细胞减少症,骨髓中有 4% 的 Eb。 和他的兄弟姐妹一样,他的 eADA 活性增加到 4.38 nmol/min/mg Hb。 再次注意到尿道下裂。 他接受了初始剂量为 2 毫克/千克/天的类固醇治疗,使 Hb 水平恢复正常。 他现在 32 岁,仍在接受每天 10 毫克的类固醇治疗。 在最后一次随访时,他表现出中度大细胞性贫血(MCV 为 105.9 fL),Hb 水平为 108 g/L,网织红细胞为 38.6 × 109/L,同时白细胞和血小板计数正常(表 2) .
DBA 的临床和生物学表现:重要信息
DBA 通常以中度至重度大细胞再生性贫血为特征。 其他细胞谱系通常是正常的,但有时,在诊断时可能会发现中性粒细胞减少症、血小板减少症,在某些情况下,甚至血小板增多症。 骨髓涂片显示其他方面正常的骨髓(无异常增生和正常细胞结构)中的红细胞减少症(<5% 的红细胞前体细胞)或骨髓活检显示的纯发育不良性贫血可以确诊。 必须进行骨髓检查以避免误诊。 成红细胞减少是红细胞在爆发形成单位-红细胞和集落形成单位-红细胞祖细胞阶段之间分化受阻的结果。 其他生物学特征包括 eADA 活性增加,在 90% 的未输血 DBA 患者中该活性升高,并且持续存在 胎儿红细胞生成的特征(高 HbF 百分比)。 它们都应在诊断时在输血前或输血后至少 3 个月进行测量(表 3)。 在 50% 的 DBA 患者中,各种畸形主要发生在头部区域和四肢,典型但罕见的三指拇指(表 4)。
表3:DBA的诊断标准(来自国际共识会议)
| 诊断标准 | 年龄小于 1 岁 |
| 无其他明显血细胞减少的大红细胞性贫血 | |
| 网织红细胞减少症 | |
| 正常的骨髓细胞结构,缺乏红细胞前体 | |
| 支持标准 | |
| 主要的 | “经典”DBA 中描述的基因突变 |
| 阳性家族史 | |
| 次要的 | 升高的 eADA 活动 |
| “经典”DBA 中描述的先天性异常 | |
| HbF 升高 | |
| 没有其他遗传性骨髓衰竭综合征的证据 |
表4:Willig 等人在 DBA 中发现的畸形
回到本文的病例,对先证者 (UPN#1447) 和她的姐姐 (UPN#1821) 以及 2 个兄弟 (UPN#1822 和 UPN#1213) 进行的分子筛检在供体附近发现了一个杂合的 4 碱基对缺失 RPS19 基因外显子 2 的剪接位点 (NM_001022.3: c.71 + 3_71 + 6del; p.?)。 在推定的 TEC 案例 (UPN#1447) 中确实发现了这种变体,它应该足以排除 TEC。 这种等位基因变异在非贫血(Hb 121 g/L;78.4 × 109/L 网织红细胞)母亲中发现,被认为是所谓的沉默表型,是 DBA 的另一个显着特征。 此外,母亲表现出正常的 MCV (96 fL) 和升高的 eADA 水平 (4.21 nmol/min/mg Hb)。 沉默的 DBA 表型个体可能是 DBA 先证者的父母或兄弟姐妹:他们没有贫血,但可能表现出大红细胞症、升高的 eADA 和/或核糖体蛋白 (RP) 基因突变。 具有“沉默表型”的患者有患 DBA 并发症(如恶性肿瘤)的风险,并且可能会将致病基因遗传下去,正如该家族中所证明的那样:母亲将该变异传给了她的所有 4 个孩子,从而加强了该家族中的显性遗传。 该家族已有2名第三代成员确诊为DBA。 UPN#1822 在 23 周 +3 天时经历了子宫内胎儿丢失 (UPN#1660),女性无畸形胎儿表现出严重的宫内发育迟缓(体重:436.5 g,介于第 5 个百分位数和第 10 个百分位数之间;头围: 18 cm,第 5 个百分位数;身高 [顶点/尾骨]:20⊊cm,第 50 个百分位数为 23 周闭经)伴羊水过少但无胎儿水肿。 有趣的是,胎盘显示出血管狭窄的缺陷,并且一些绒毛血管化。 然而,未发现成红细胞减少症。 已在该胎儿中鉴定出相同的等位基因变异。 我们假设与 DBA 相关的主要生长迟缓可能是胎儿在子宫内死亡的原因(表 5)。 胎儿水肿是 DBA 的一个特征,其发生率可能被低估了。 自从基因解码描述了第一例因 RPS19 基因突变导致的偶发性胎儿流产病例后,通过基因检测随后发现了更多的胎儿病例,这些病例主要与 RPS19 基因相关,也与 RPL15 基因相关。 正如佳学基因案例之前所述,这些发现进一步证实了这一点 , 怀孕期间可能发生主要并发症,以及在怀孕期间使用推定的阿司匹林治疗对母亲及其后代进行随访的重要性,以预防案例中提到的胎盘血管并发症。
表 5:DBA 的表征模式
| 中位年龄为 2 个月;新生儿(占病例的 16%);胎儿水肿 |
| 再生性通常为大红细胞性贫血 |
| 正常细胞骨髓中的成红细胞减少症 |
| 畸形,特别是腭裂、拇指畸形、心脏和泌尿生殖道畸形 |
| 身材矮小,包括宫内发育迟缓 |
| eADA 提升 |
| 胎儿红细胞生成特征(6 个月后 HbF 百分比升高) |
| 妊娠并发症 |
| 恶性肿瘤 |
| 非常罕见的再生障碍性贫血 |
除了 UPN#1822,UPN#1821 还有 5 个后代,其中有双胞胎(UPN#779、UNP#1114、UPN#1264、UPN#1573、UNP#1574)(图 1)。 产前诊断被拒绝,在 5 名儿童中,只有 1 名男孩患有 DBA,并且在 RPS19 基因中携带相同的家族性等位基因变异。 UNP#1264 出生时正常妊娠时间为 40 周,月经正常,但结膜和皮肤苍白、心动过速和收缩期心脏杂音是由于 120 g/L 的贫血(正常值 >140 g/L),下降到 出生后第 18 天为 75 g/L。 他被输注了 70mL 红细胞。 从那以后,他每 3 到 4 周定期接受一次输血。 他没有表现出任何先天性异常。 1 年后按建议开始类固醇治疗,但没有反应。 开始使用地拉罗司进行螯合治疗,但由于肝毒性不得不中断。 7 天中有 5 天每晚输注 10 小时以上的去铁胺治疗 (1000 mg/d) 开始控制铁过载。 考虑进行干细胞移植,并对兄弟姐妹进行了已知的家族性等位基因变异检测,但在他们中均未发现。 不幸的是,它们都不是 HLA 相同的。 他目前 6 岁,最近接受了有效匹配的无关供体的移植。
DBA 的治疗:
2021 年的 DBA 治疗仍然依赖于长期输血(Hb 浓度保持 >90 g/L),当铁蛋白水平 >500 µg/L 时开始最佳铁螯合疗法,或使用皮质类固醇治疗。皮质类固醇( prednisone 或 prednisolone) 只能在出生后的第一年引入,以实现最大生长,因为身材矮小是畸形综合征的一部分。 皮质类固醇以 2 mg/kg/d 的剂量引入,其疗效通常在治疗开始后 2 周开始显现,表现为网织红细胞计数和 Hb 水平增加。 如果 1 个月后对类固醇治疗没有反应,则没有理由继续这种治疗。 如有效,皮质类固醇应逐渐减量至能维持Hb水平在90g/L以上的最小剂量。 皮质类固醇的最大连续剂量必须<0.3‰mg/kg/d(在输血困难或危险的国家<0.5‰mg/kg/d)。 在皮质抵抗或皮质依赖(>0.3 [或>0.5‰mg/kg/d])的情况下,铁螯合输血是目前唯一的替代治疗选择,造血干细胞移植 (HSCT) 排除了 DBA 或沉默表型的 HLA 相同兄弟姐妹或与有效匹配的 (10/10) 无关供体。HSCT 最好在 5 岁之前进行,当然 <10 年避免 HSCT 并发症。 HSCT 也适用于克隆进化患者。造血干细胞的替代来源应被视为实验方法,但可能适用于克隆进化患者。 HSCT 可治好贫血并预防骨髓增生异常综合征 (MDS) 和白血病的风险,但仍需要仔细监测 DBA 患者的移植后实体瘤风险。
最后,已发现亮氨酸对少数 DBA 患者有效。基因疗法及其他新的治疗选择可能是最有希望的选择。最近,提出了一些创新疗法 例如钙调蛋白抑制剂和二甲双胍,转化研究可能会出现其他新的治疗选择(使用三氟拉嗪的试验 NCT03966053)。
DBA 致病基因鉴定基因解码
在大约 70% 至 80% 的受 DBA 影响的病例中发现等位基因变异在 RP 中始终处于杂合状态。已鉴定出 20 个 RP 基因中的突变(图 2)。 在佳学基因收集的临床病例中发现的最频繁突变的基因是 RPS19 基因,它占全球 DBA 病例的 25%。 在大约 20% 的 DBA 病例中发现了这些 RP 基因的大量缺失,主要是在 RPS17 、RPL35a 和 RPS19 基因。 因此,至少 70% 的 DBA 患者携带等位基因变异,包括仅 8 个 RP 基因的大缺失,即 RPS19、RPL5、RPS26、RPL11、RPL35a、RPS10、RPS24、 和 RPS17(图 2)。 迄今为止,尚未报道 DBA 患者的多种致病性 RP 突变。 表型/基因型相关性并不明显,尽管已经确定携带 RPS19 基因突变的 DBA 患者与其他患者相比表现出较少的畸形,但似乎表现出更严重的血液学表型并且经常通过输血进行治疗。 中性粒细胞减少症是更频繁地与 RPL35a、心脏异常与 RPS24、腭裂与 RPL5 和拇指异常与 RPL1136 基因突变相关。 已发现其他 RP 基因在受 DBA 影响的患者的一小部分中发生突变,每个影响 <1% 的 DBA 队列(图 2)。 DBA 中涉及的 RP 基因突变与核糖体 RNA (rRNA) 成熟缺陷有关,这被认为是 DBA 疾病的标志性特征,基因解码证据证实了意义不明的等位基因变异的致病性。 DBA 样疾病导致贫血伴成红细胞减少和一定程度的类固醇反应,但 rRNA 成熟没有缺陷。 这些 DBA 样疾病与 EPO 和 GATA基因突变有关。 DBA和DBA样疾病构成DBA综合征。 尽管成红细胞减少症和一定程度的类固醇反应,与 CECR1 或 ADA2 基因突变相关的 ADA2 缺陷不被认为属于 DBA 综合征(图 2)。
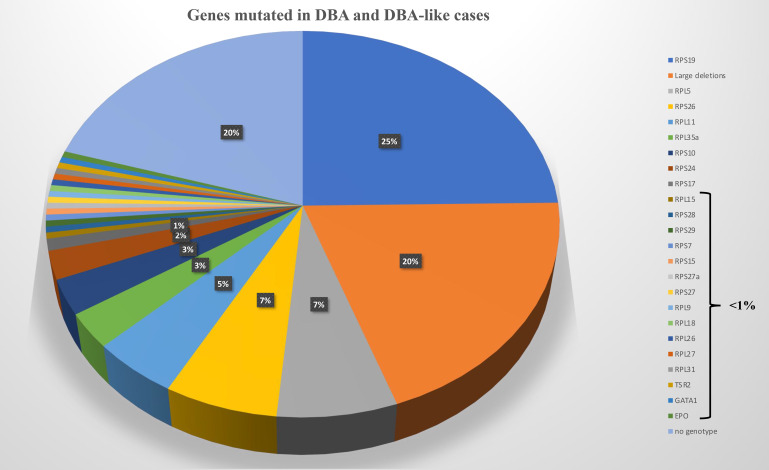
基因在 DBA 和类似 DBA 的病例中发生突变
佳学基因收录的这一DBA 家族已受到恶性肿瘤的影响。 代表 DBA 沉默表型的母亲被诊断出患有迅速致命的乳腺癌。 最年长的后代 UNP#1447 携带家族性 RPS19 等位基因变异但从不需要任何治疗,在 36 岁时被诊断出患有直肠腺癌,并在治疗 16 个月后去世。 这些观察表明识别 DBA 患者(而不是将他们误诊为 TEC)很重要,并且在随访中应考虑 DBA 患者所谓的沉默 DBA 表型,特别是考虑到恶性肿瘤的风险。 最近的一项研究确定,在美国国家癌症研究所收收录的DBA病例中,患有 DBA 的非移植患者患恶性肿瘤的风险为 5%,任何恶性肿瘤的观察/预期比率为 4.8,结肠癌为 44.7,肺癌为 9.4 癌症,成骨肉瘤 42.4,MDS 352,急性髓性白血病 28.8。然而,与其他遗传性骨髓衰竭综合征相比,DBA 的观察/预期比率较低。迄今为止,尚无 MDS/急性髓性白血病和实体瘤筛查的指南,除了最近关于结直肠癌的初步建议,但这可能是必要的,目前正在 DBA 合作小组中进行讨论。
总之,DBA 是一种引人入胜且复杂的红系疾病,正如佳学基因所收录的这一DBA 家族研究所示。 佳学基因致力于研究DBA 的分子基础、疾病的病理生理学以及开发和寻求新的治疗选择方面正在取得进展。在持续的研究的影响下,些进展将在未来几年为 DBA 患者提供更好的临床管理。
- 上一篇:没有了
- 下一篇:【佳学基因检测】Léri-Weill综合征基因检测基因解码(LWD)
- 【佳学基因检测】唇裂、腭裂的基因检查与检测分析...
- 【佳学基因检测】怎么筛查唇腭裂?...
- 【佳学基因检测】唇裂与腭裂基因检测...
- 【佳学基因检测】基因检测说明唇腭裂是否是因为怀孕期间吃了什么东西?...
- 【佳学基因检测】唇腭裂基因检测可以找到畸形发生的原因吗?...
- 【佳学基因检测】常见骨科基因检测项目,这些疾病会遗传!...
- 【佳学基因检测】哪些骨头病会遗传,如何做基因检测才能阻断遗传,帮助治疗?...
- 【佳学基因检测】骨科遗传性疾病基因检测...
- 【佳学基因检测】什么时候会诊断为疑似戈谢病?怎么做基因检测分析?...
- 【佳学基因检测】软组织和骨肿瘤基因检测分子病理学共识与标准...
- 【佳学基因检测】骨科疾病为什么要做染色体基因检测?检测结果如何指导遗传阻断?...
- 【佳学基因检测】用病例说明早衰症基因检测是怎么做的?...
- 【佳学基因检测】骨穿基因检测是什么?...
- 【佳学基因检测】 骨科检查项目及费用...
- 【佳学基因检测】骨科基因检测项目有哪些?...
- 【佳学基因检测】基因检测如何找到骨关节炎效果基因?...
- 【佳学基因检测】脊柱侧弯基因检测细分型、好治疗!...
- 【佳学基因检测】并指症基因检测,怎样才能知道如何阻断遗传?...
- 【佳学基因检测】多了一个指头是病吗,是否遗传,基因检测给答案!...
- 【佳学基因检测】遗传性佝偻病基因检测,优质机构推荐...
- 【佳学基因检测】遗传性矮小症基因检测...
- 【佳学基因检测】骨代谢性疾病基因检测...
- 【佳学基因检测】先天性软骨病基因检测及其对治疗的帮助作用...
- 【佳学基因检测】揭秘“瓷娃娃”成骨不全症,基因检测如何给予帮助?...
- 【佳学基因检测】成骨不全症基因检测(Osteogenesis imperfecta)...
- 【佳学基因检测】骨质疏松需要做基因检测吗?...
- 【佳学基因检测】抗骨质疏松药用药安全基因检测:怎么做?...
- 【佳学基因检测】大理石骨病基因检测如何帮助治疗...
- 【佳学基因检测】骨石症基因检测如何选择可信赖的机构...
- 【佳学基因检测】硬骨症基因检测及其治疗方案...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
