












一个"低钙"了16年的男孩:CASR基因突变如何改写常染色体显性遗传性低钙血症的诊治之路
从新生儿期反复抽搐,到基因检测锁定病因,再到新型长效PTH类似物带来转机——一例真实病例背后的基因科学
核心提示:常染色体显性遗传性低钙血症1型(ADH1)是一种由CASR基因功能获得性(激活性)突变引起的罕见内分泌遗传病。患者因甲状旁腺激素(PTH)分泌被过度抑制,长期处于低血钙、高血磷状态,若按常规低钙血症思路"补钙",反而可能加重肾脏损害。近期波兰罗兹医科大学团队在《Frontiers in Endocrinology》发表的一例青少年病例,清晰展示了基因检测在这类"疑难低钙血症"诊断中的决定性作用。
一、被忽视的可能性:低血钙不一定是"缺钙"
提到低血钙,多数人第一反应是"补钙"。但在临床上,有一类患者的低血钙恰恰源于身体对钙"过度敏感"——他们的甲状旁腺激素分泌被自身的调节系统异常压制了。这类疾病的核心分子机制,指向了一个名为CASR(钙敏感受体)的基因。
CASR基因编码的钙敏感受体,广泛表达于甲状旁腺和肾脏,负责感知血液中钙离子浓度、进而调控PTH分泌和肾脏对钙的重吸收。当CASR基因发生功能获得性(激活性)突变时,受体会在血钙尚处于正常甚至偏低水平时就"误判"为钙已经足够,从而过度抑制PTH分泌,并促使肾脏排出更多的钙——这就是常染色体显性遗传性低钙血症1型(ADH1,OMIM 601198)的发病机制。
与之相对,CASR基因的功能缺失性突变会导致完全相反的疾病——家族性低尿钙性高钙血症(FHH);而新生儿严重原发性甲状旁腺功能亢进(NSPHT)也与CASR相关。同一个基因,突变方向不同,疾病表现截然相反,这正是遗传病诊断中"基因型-表型精准对应"的典型例子。
二、真实病例:16年求医路,直到基因检测给出答案
病例中的男孩,出生仅6周便因泌尿系统感染住院,意外查出严重低血钙、高血磷、PTH几乎测不出,被诊断为先天性甲状旁腺功能减退症。此后十余年间,他反复出现手足麻木、肌肉痉挛、抽搐,每年因此住院1至2次,尤其在感染、运动后或天气炎热时症状加重。
更棘手的是,孩子同时合并尿钙排泄增多——1岁半时超声就已发现双肾多发肾钙质沉积和肾结石,后来还出现了脑基底节钙化和早发性白内障。2010年,他曾接受同种异体甲状旁腺细胞移植手术,但移植未能发挥功能。常规治疗(骨化三醇类药物+补钙+补镁)虽然维持多年,却始终无法真正控制病情,血磷持续偏高,钙磷乘积居高不下——这是一个提示肾脏及软组织异位钙化风险的重要指标。
直到2015年,孩子7岁时,基因检测终于揭示了病因:CASR基因发生了一个杂合错义突变 c.2504C>A(p.Ala835Asp),且父母均未携带该变异,属于新发(de novo)突变。这个位于第7号外显子、第七跨膜结构域细胞外环3的位点,恰好是ADH1激活性突变的"热点区域",此前已有功能实验证实同一位置的氨基酸替换会导致钙信号通路在低钙浓度下被异常激活。
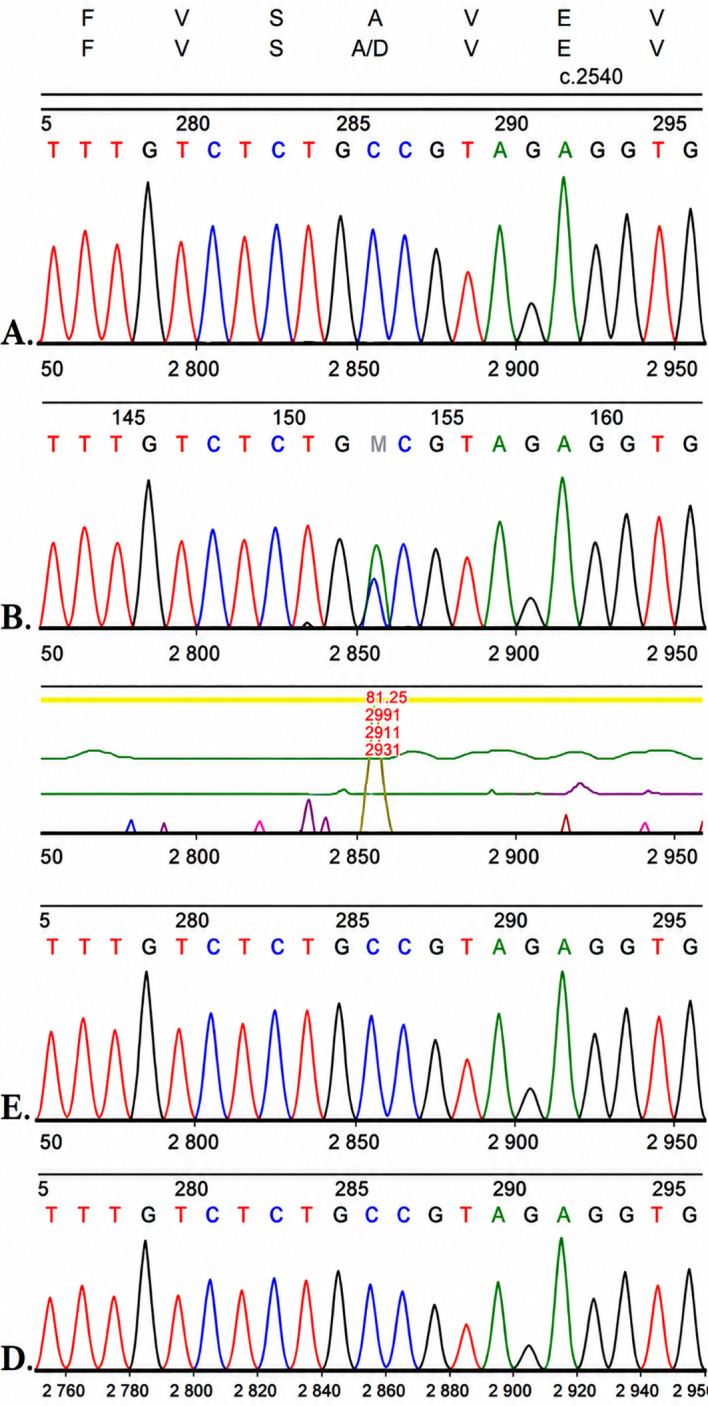
三、为什么基因检测是ADH1诊断的"金标准"
在没有颈部手术史的低血钙患者中,遗传学评估应作为常规检查手段之一,尤其是儿童患者。ADH1的鉴别诊断范围并不窄——包括GNA11基因突变导致的ADH2型(临床表现相似但通常没有明显高尿钙和低镁血症)、GCM2/PTH基因突变、DiGeorge综合征(22q11.2缺失)、HDR综合征(甲状旁腺功能减退-耳聋-肾发育不良综合征),以及自身免疫性或低镁血症导致的功能性甲状旁腺功能减退。
单靠临床表现,很难在这些疾病之间做出确定性区分;而基因检测能够直接定位致病基因及具体突变位点,结合家系验证(父母、同胞是否携带同一变异),可以明确:
| 诊断价值 | 意义 |
|---|---|
| 明确致病基因与突变类型 | 区分激活性/失活性突变,判断疾病走向(低钙 vs 高钙) |
| 判断遗传方式(新发 vs 遗传) | 评估家族再发风险,指导生育决策与产前/植入前诊断 |
| 避免误诊误治 | 防止"经验性补钙"加重高尿钙、肾钙质沉积甚至肾功能损害 |
| 指导精准治疗方案选择 | 为靶向CaSR的新型药物(如钙受体拮抗剂)临床决策提供依据 |
值得一提的是,本例中,虽然孩子的父母单次血钙检测也略偏低,但基因检测结果排除了他们携带同一致病变异——这说明不能仅凭一次生化指标下诊断结论,基因检测才是确认致病突变来源、判断是否为新发突变的关键依据。
四、治疗新进展:从"经验补钙"到精准干预
ADH1的传统治疗以骨化三醇类药物联合补钙为主,但这一疗法存在天然矛盾:升高血钙的同时会显著加重尿钙排泄,长期可能诱发肾钙质沉积、肾结石乃至肾功能损害及其他组织异位钙化。因此国际指南建议,ADH1患者的目标血钙应维持在正常范围下限以下,仅以缓解神经肌肉症状为治疗终点,而非追求血钙"达标"。
病例中的团队在常规治疗效果不佳的情况下,尝试了一种新型长效甲状旁腺激素类似物(palopegteriparatide),这是一种通过皮下注射持续、稳定释放PTH(1-34)的药物,目前已获批用于成人慢性甲状旁腺功能减退症治疗,但此前尚无儿童ADH1患者使用的公开报道。
经过审慎的剂量滴定(从9μg/天逐步调整至7.5μg/天),并配合利尿剂减量,患者在治疗12个月后实现了:抽搐症状完全消失、钙磷乘积由治疗前的56.8 mg²/dL²降至约52.2 mg²/dL²,并成功停用了骨化三醇和钙剂补充。这一结果为常规治疗失败的儿童ADH1患者提供了新的治疗思路,尽管其长期安全性仍需更多研究验证。
与此同时,针对CaSR本身的"钙受体拮抗剂"(calcilytics,如encaleret)也正处于临床试验阶段,这类药物直接作用于突变受体、从源头纠正信号异常,代表了ADH1治疗未来更具针对性的方向。可以预见,随着对CASR基因功能认识的不断深入,"病因导向"的精准治疗将逐步取代"对症补钙"的传统模式。
五、这个病例给我们的启示
这例长达16年的诊治历程提示我们:面对反复发作、常规治疗效果不佳的低血钙、高血磷或不明原因的手足抽搐、肾结石、脑内钙化等表现,及时进行致病基因鉴定,往往是打破"经验性治疗僵局"的关键一步。基因检测不仅能明确诊断、避免不恰当的补钙治疗带来的肾脏损害,更能为家庭的生育决策——包括是否需要考虑遗传咨询、产前诊断或植入前遗传学检测——提供科学依据。
佳学基因检测专注于致病基因鉴定,通过系统化的基因检测流程,帮助疑难低钙血症、不明原因抽搐、反复肾结石等罕见遗传病患者及家庭准确查找疾病原因,为后续的精准治疗方案选择与生育干预决策提供坚实的科学依据——让每一个家庭在漫长的求医路上,多一份信心,多一份安心,多一份放心。
本文根据 Zygmunt A, et al. Case Report: Palopegteriparatide as a novel therapeutic option in pediatric autosomal dominant hypocalcemia type 1. Front Endocrinol. 2026;17:1837966(开放获取文献,CC BY许可)整理改写,用于科普目的,不构成具体诊疗建议。
佳学基因检测
致病基因鉴定 · 基因检测准确查找疾病原因 · 为精准治疗和生育干预提供信心、安心、放心
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:基因检测)
