介绍
许多不同癌症的基因研究已经确定了少数必须突变或改变以促进恶性细胞生长的基因[ 1 ]。癌细胞的两个主要特性,不受控制的细胞生长和侵入其他组织的能力,是这些遗传和表观遗传变化的结果。遗传交替包括基因突变、基因组不稳定性、杂合性丢失 (LOH) 和基因拷贝数变异 (CNV)。相比之下,表观遗传变化包括组蛋白修饰、DNA 甲基化和印记丢失 (LOI)。这些修饰在不改变潜在核苷酸序列的情况下调节基因表达[ 2-4 ]。
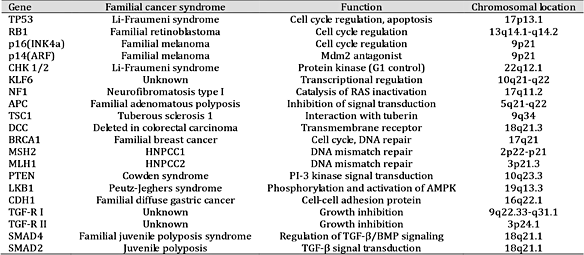
一般来说,癌症相关基因可以分为两大类,原癌基因和肿瘤抑制基因(TSG)。原癌基因通常参与促进细胞生长的途径。当这些基因被突变或改变激活时,它们会导致正常细胞癌变。原癌基因的突变通常在自然界中占主导地位,这些基因的突变版本被称为癌基因 [ 5 ]。TSGs被认为是另一种关键基因,参与DNA损伤修复、抑制细胞分裂、诱导细胞凋亡和抑制转移。因此,TSGs 功能的丧失会导致癌症的发生和进展 [ 6]。先前的研究表明,一个 TSG 副本足以控制细胞增殖。因此,TSG 的两个等位基因必须长久失活或丢失才能导致肿瘤发展 [ 7 ]。此外,根据具体情况,一些基因既可以作为原癌基因也可以作为 TSG。TSG 大致分为五种类型: 1) 编码细胞内蛋白质的基因,控制进入细胞周期的特定阶段(例如 pRB 和 p16)[ 8 ];2) 编码受体或信号转导的分泌激素或抑制细胞增殖的发育信号的基因 [例如,转化生长因子 (TGF)-β 和腺瘤性结肠息肉 (APC)] [ 9]; 3) 编码检查点控制蛋白的基因,这些蛋白触发细胞周期停滞以响应 DNA 损伤或染色体缺陷[例如,乳腺癌 1 型易感蛋白 (BRCA1)、p16 和 p14] [ 10 ];4) 编码诱导细胞凋亡的蛋白质的基因(例如,p53)[ 11 ];5) 编码参与修复 DNA 错误的蛋白质的基因 [例如,p53 和 DNA 错配修复蛋白 2 (MSH2)] [ 12 ]。
TSG失活是导致癌症发展的常见机制。分子研究表明,TSGs 的失活通常与细胞遗传学上无法检测到的微缺失有关,这些微缺失是通过显示在肿瘤抑制基因座内或附近的多态性标记的 LOH 来确定的 [ 13 ]。TSG 中的种系突变是大多数已知的可遗传癌症形式的原因,因为一个等位基因的体细胞失活通常与正常发育相容 [ 14 ]。此外,启动子甲基化也有助于 TSG 的失活 [ 15 , 16 ]。迄今为止发现的大多数 TSG 都遵循上述 Knudson 范式(表1)。这些 TSG 在细胞水平上是隐性的,并且两个等位基因都被癌症中的甲基化删除、突变或沉默。然而,对人类肿瘤标本的研究越来越多的证据表明,通过蛋白酶体降解、异常细胞定位和转录调控等细胞机制使 TSG 功能失活也可能导致肿瘤发生。本综述概述了不遵循经典 Knudson 两次打击假设的已知 TSG 的失活机制。这些 TSG 的分子分析可能会揭示特定癌症亚类的新靶点。
表格1:抑癌基因的位置和功能
泛素-蛋白酶体降解途径
必须严格调节由 TSG 和癌基因编码的蛋白质的细胞水平,以防止癌变和恶性进展。TSG 产物的水平通常由泛素-蛋白酶体途径控制,这是一种特定的细胞蛋白水解机制。E3泛素连接酶通过与泛素激活酶E1和泛素结合酶E2的协同作用,催化其特定蛋白底物的多泛素化,然后修饰的底物被26S蛋白酶体降解[ 17 ]。由于泛素化-蛋白酶体途径功能障碍或 E3 连接酶异常表达导致的 TSG 产物降解增强可能与肿瘤发生有关。例如,下面讨论了通过泛素-蛋白酶体降解使 TSG 功能失活。
泛素-蛋白酶体降解使 p53 TSG (TP53) 失活
p53 TSG (TP53) 在大多数癌症中失活 [ 18 , 19 ]。它负向调节细胞周期并参与基因组稳定和血管生成 [ 18 , 19 ]。在人类癌症中经常报道纯合缺失 (HD)、LOH、点突变和/或甲基化导致TP53失活(表2-9)。除了这些遗传失活机制外,细胞 p53 水平还通过泛素化介导的降解进行调节 [ 20]。许多含有环指结构域的 E3 连接酶,包括小鼠双分钟 2 同源物 (MDM2)、MDM4、疱疹病毒相关泛素特异性蛋白酶 (HAUSP)、组成型光形态发生 1 (COP1)、Pirh2 和 ARF-BP1,可以泛素化第 53 页 [ 21 - 25 ]。然而,MDM2 似乎是主要的调节因子,因为MDM2缺乏症的致死性可以通过TP53的损失来挽救[ 21 , 26 ]。通过与 MDM4 的异二聚化,MDM2 对 p53 的 E3 连接酶活性显着增加 [ 21 , 26 ]。在大约 10% 的肿瘤中观察到MDM2的基因扩增[ 21 ,26 ],其中大部分保留了野生型TP53。在一些肿瘤细胞中,即使在没有MDM2基因扩增的情况下,也会出现 MDM2 的过表达 [ 27 ]。此外,在大约 10% 的所有肿瘤和 65% 的视网膜母细胞瘤中发现了MDM4的过表达 [ 21 , 28 ]。病毒也可能促进 p53 的降解。人乳头瘤病毒 E6 蛋白与 E3 泛素连接酶的 E6 羧基末端 (HECT) 结构域同源形成复合物,导致 p53 泛素化和降解 [ 21 , 29 , 30]。这些发现与泛素-蛋白酶体途径通过p53功能丧失促进肿瘤发生的观点一致。
表 2。
肿瘤抑制基因在白血病和脑癌中的表达和失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的
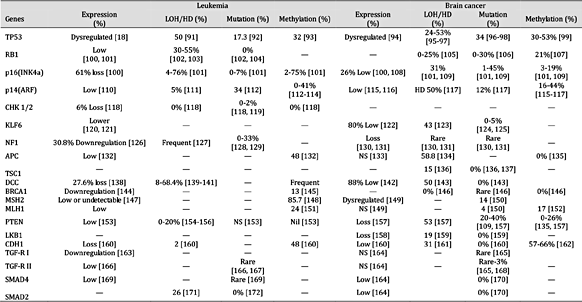
表3。
抑癌基因在头颈癌和肺癌中的表达及失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的
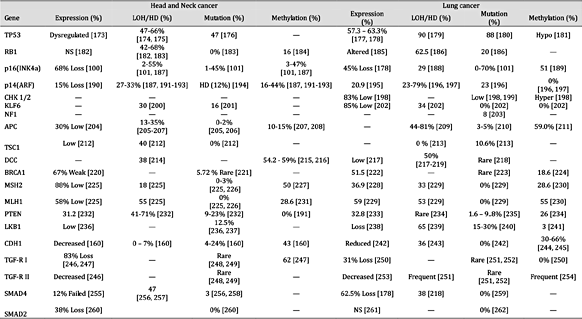
表 4。
肿瘤抑制基因在癌症中的表达和失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的
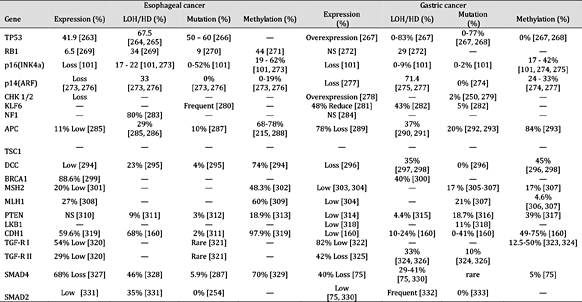
表 5。
肿瘤抑制基因在癌症中的表达和失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的
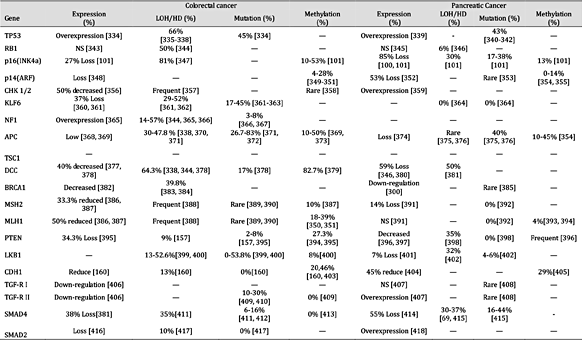
表 6。
肿瘤抑制基因在癌症中的表达和失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的
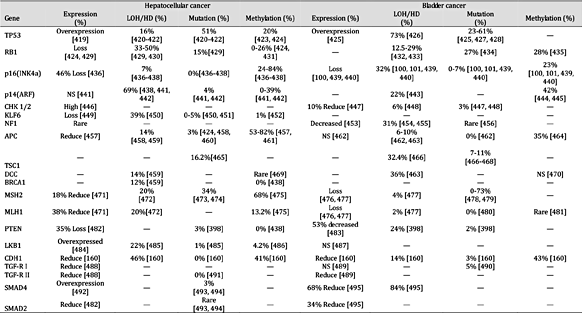
表 7。
肿瘤抑制基因在癌症中的表达和失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的
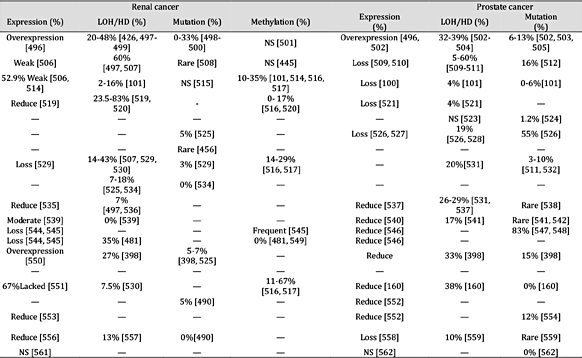
表 8。
肿瘤抑制基因在乳腺癌和卵巢癌中的表达和失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的
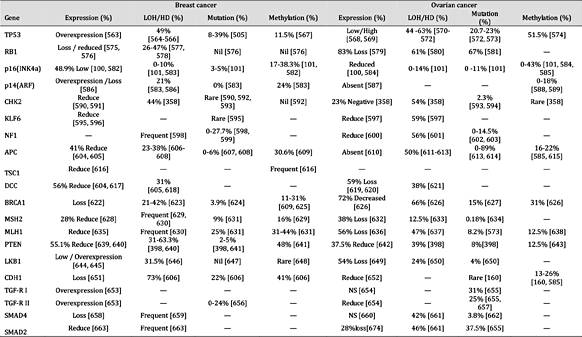
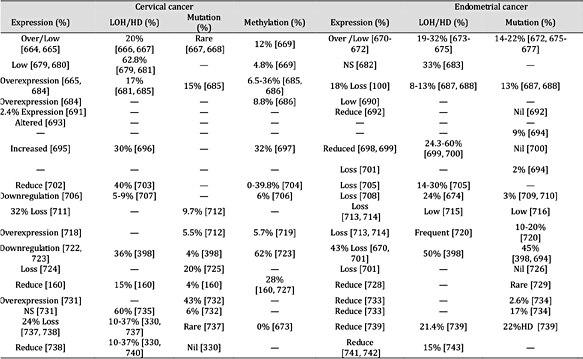
表 9。
抑癌基因在宫颈癌和子宫内膜癌中的表达及失活机制。注:“—”未找到报告;“HD”纯合性缺失;“NS”无显着差异;平均百分比是使用总病例除以阳性病例计算的