












【佳学基因检测】肿瘤个体化治疗检测技术指南(试行)——国家标准
前言
肿瘤的个体化治疗基因检测已在临床广泛应用,实现肿瘤个体化用药基因检测标准化和规范化,是一项意义重大的紧迫任务。本指南从诊断项目的科学性、医学实验室检测方法的准入、样本采集至检测报告发出的检测流程、实验室质量高效体系四个方面展开了相关论述,使临床医生能够了解所开展检测项目的临床目的、理解检测结果的临床意义及对治疗的作用;医学实验室为患者或临床医护人员提供及时、正确的检验报告,并为其提供与报告相关的咨询服务。检测技术的标准化和实验室准入及质量高效对临床和医学实验室提出了具体的要求,以最大程度的高效检测结果的正确性。
本指南是参考现行相关的法规和标准以及当前认知水平下制定的,随着法规和标准的不断完善,以及肿瘤个体化治疗靶点基因的不断发现,本技术规范相关内容也将进行适时调整。
本指南起草单位:中国医学科学院肿瘤医院分子肿瘤学国家重点实验室、苏州生物医药创新中心,经国家卫生计生委个体化医学检测技术专家委员会、中国抗癌协会相关专业委员会、中华医学会检验医学分会、中华医学会肿瘤学分会的专家修订。
本指南起草人:詹启敏、曾益新、王珏、姬云、钱海利、李晓燕、孙石磊
1. 本指南使用范围
本指南由国家卫生计生委个体化医学检测技术专家委员会制定,是国家卫生计生委个体化医学检测指南的重要内容,旨在为临床分子检测实验室进行肿瘤个体化用药基因的检测提供指导。本指南的主要适用对象为开展个体化医学分子检测的医疗机构临床分子检测实验室。
2. 简介
肿瘤个体化治疗以疾病靶点基因诊断信息为基础,以循证医学研究结果为依据,为患者提供接受正确治疗方案的依据,已经成为现代医学发展的趋势。临床研究证实,通过检测肿瘤患者生物样本中生物标志物的基因突变、基因SNP分型、基因及其蛋白表达状态来预测药物疗效和评价预后,指导临床个体化治疗,能够提高疗效,减轻不良反应,促进医疗资源的合理利用。
3. 标准术语和基因突变命名
3.1标准术语
1)基因(Gene):是遗传物质的最小功能单位,是指具有一定生物学意义的一段DNA。
2)突变(Mutation):是细胞中DNA核苷酸序列发生了稳定的改变。
3)融合基因(Fusion gene):是指两个基因的全部或一部分的序列相互融合为一个新的基因的过程。融合基因的表达产物为融合蛋白。
4)基因表达(Gene Expression):是基因中的DNA序列生产出蛋白质的过程。步骤从DNA转录成mRNA开始,一直到对于蛋白质进行翻译后修饰为止。
5)基因扩增(gene amplification):为一特异蛋白质编码的基因的拷贝数选择性地增加而其他基因并未按比例增加的过程。
6)DNA甲基化(DNA Methylation):为DNA化学修饰的一种形式,将甲基添加到DNA分子上,例如在胞嘧啶环的5’碳上。DNA甲基化能在不改变DNA序列的前提下,将DNA甲基化状态遗传至下一代细胞或个体。
7)聚合酶链反应(PCR):是一种体外扩增特异DNA片段的技术。利用DNA在体外摄氏高温时变性会变成单链,低温时引物与单链按碱基互补配对的原则结合,再调温度至DNA聚合酶最适反应温度,DNA聚合酶沿着磷酸到五碳糖(5′-3′)的方向合成互补链。
8)质控品:是含量或成分已知的处于与实际样本相同的基质中的特性明确的物质,这种物质通常与其他杂质混在一起,专门用于质量控制目的的样本或溶液。
3.2 基因突变命名
人类基因组突变学会(HGVS)已建立系统的基因突变命名方法。具体基因突变命名方法可查阅网站http://www.hgvs.org/mutnomen/index.html。HGVS基因突变命名指南根据需求不断更新。本文以2011年8月更新版本为准。
当描述某一序列改变时,其前缀表明其参考序列类型。例如“g.”表示基因组序列,“c.”表示cDNA序列,“m.”表示线粒体DNA序列,“r.”表示RNA序列,“p.”表示蛋白序列。在数据库中的收录号以及版本号应当在实验记录报告中列出,当两种突变在反式(in trans)中检测到,则用方括号表示。例如,CF突变为杂合性突变(508号苯丙氨酸缺失和1303号天冬酰胺被赖氨酸替代),则在DNA水平规范描述方式为c.[1521_1523delCTT]+[3909C>G]。
3.3 参考序列
美国国立生物技术信息中心(NCBI)收录的参考序列编码具有权威性及唯一性。其中前缀“NM_”表示为mRNA序列,“NP_”表示多肽序列,“NG_”表示基因组序列。基因组参考序列应列出完整基因序列,包括5’以及3’非编码区(UTR)。当使用某段编码DNA参考序列描述突变时,应选择合适的转录体,且转录体的起始转录点应当明确,例如选择最常见的转录体,或者是已知的最大转录体,或者具有组织特异性的编辑转录体。当某一参考序列具有多种转录方式时,选择NCBI数据库里注释最全面的版本。
3.4 各类变异
3.4.1 DNA序列变异术语规范
DNA核苷酸用大写字母A(腺嘌呤)、C(胞嘧啶)、G(鸟嘌呤)以及T(胸腺嘧啶)来表示。用正链来表示DNA序列。当DNA序列改变时,以相应核苷酸所在位置及相应字母来描述。“>”符号表示“从某一变化至另一”。在描述突变方式时,数字、字母、箭头、上标以及下标之间不应出现空格。
3.4.2 RNA序列变异术语规范
RNA序列以小写字母a(腺嘌呤)、c(胞嘧啶)、g(鸟嘌呤)、u(尿嘧啶)进行描述。RNA序列改变描述方式与DNA相类似。具体术语可参阅HGVS网站(http://www.hgvs.org/mutnomen/index.html)。
3.4.3蛋白质序列变异术语规范
蛋白质序列改变通常以单个字母或三个字母(第一个字母大写)来描述。尽管单个字母描述氨基酸明确无误,但是由于三联密码子相对于其编码的氨基酸存在冗余性,具体给出发生突变的三联体密码子可以更清楚地描述氨基酸改变方式。例如,用来描述氨基酸的A(丙氨酸)、C(半胱氨酸)、G(甘氨酸)以及T(苏氨酸)可能会与核苷酸字母A(腺嘌呤)、C(胞嘧啶)、G(鸟嘌呤)以及T(胸腺嘧啶)相混淆。
3.4.4错义突变及无义变异术语规范
由于三联体密码子的简并性,多个位点核苷酸的改变可能不影响最终氨基酸序列。因此,应该分别从DNA水平和氨基酸水平描述突变。从DNA水平对某一突变位点的描述方式包括碱基位点,正常碱基,“>”符号,突变碱基。例如,某一蛋白第551号氨基酸残基由G(甘氨酸)突变为D(天冬氨酸),从DNA水平描述即c.1652G>A。
在氨基酸水平,由于错义突变的产物以氨基酸残基位点以及表示氨基酸的单字母或三联体密码子来描述。表示方法是野生型的氨基酸、位点、突变氨基酸,三者之间不要有空格。例如,p.Gly551Asp表示该蛋白中551号甘氨酸残基(G)被天冬氨酸残基(D)所代替。无义突变表示方法与之相类似。需要指出的是“X”符号代表终止密码子。例如,p.Gly542X表示542位点的甘氨酸残基被终止密码子所代替。
3.4.5 缺失和插入术语规范
缺失和插入突变分别用前缀“del”和“ins”来表示,并注明突变位点以及碱基。例如,c.441delA表示在该DNA序列中441号位点发生A碱基缺失。c.241_243delATC表示在该DNA序列中从241号到243号缺失ATC三个碱基。
在蛋白水平,上述突变描述方式为p.Ile24del,表示该蛋白质中第24号的异亮氨酸残基发生缺失。“indels”则表示该段序列缺失的同时有片段插入。例如,234_239delAATTCGinsTA(或者234_239delinsTA)表示该DNA序列234至239号位点缺失AATTCG六个碱基,同时该段位点被新插入的TA碱基所替代。
3.4.6 移码突变术语规范
移码突变用“fs”符号来表示。“fs*#”则用于进一步描述突变类型。例如,p.His62Profs*21表示该蛋白发生移码突变,第62号氨基酸由组氨酸突变为脯氨酸并产生新的阅读框架,终止于第62号密码子下游21号密码子处。该突变也可简要描述为p.His62fs,即该蛋白从第62号密码子发生移码突变。
3.4.7 碱基重复序
HGVS推荐,核苷酸重复序列基因多态性描述时通常以一个重复序列为单元,后面加上“[重复的次数]”,如CGG[55]。当重复序列的次数在一个范围之内时,需要在小括号“()”中标注出可能的最少的和最高的重复次数,如某个人HTT基因中发现有12个和15个CAG重复,基因水平的表示如下:c.52CAG[12]+[15],蛋白水平则表示为:p.Gln18[12]+[15]。HTT基因的重复序列范围则描述为:c.52CAG(27_35) 或 p.Gln18(27_35)。
3.4.8 遗传药理学基因型术语规范
最广泛使用的命名法描述遗传药理学基因型不同于其他遗传学基因检测,例如代谢相关基因细胞色素p450家族,人类细胞色素P450(CYP)等位基因命名委员会(http://www.cypalleles.ki.se)推荐用“*”命名,见图1。在该系统中,最常见的等位基因被指定为“* 1”。
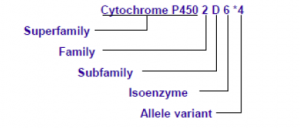
图1.细胞色素P4502D6等位基因命名规范
3.4.9 其他
对于更复杂的突变,可参考HGVS(http://www.hgvs.org/mutnomen)建议的命名规则,解决其他复杂的变异的命名。
4. 分析前质量高效
4.1 样本类型及获取
4.1.1 新鲜组织(包括手术和活检组织)
肿瘤新鲜冷冻材料可提取出最高品质的DNA、RNA。在手术现场取样的情况也比较多,但需要在显微镜下确认肿瘤细胞含量。周围炎症严重的肿瘤、黏液产生过高的肿瘤、病变中心广泛纤维化的肿瘤细胞不能采集,以免产生假阴性结果。切割后取其中一半,并利用另一半切面制作组织标本,然后进行确认。
手术切除的组织样本理想的保存方法是迅速置于液氮中,然后保存于液氮罐或-80℃冰箱,这一过程应在手术样本离体后30分钟内完成。由于组织样本通常需先进行病理学分析,在分析完成后应尽早将组织样本置于稳定剂中,避免核酸降解。
4.1.2 石蜡包埋组织(formalin-fixed paraffin-embedded,FFPE)
10%中性福尔马林固定手术切除样本,按病理学操作规范进行取材。
制作石蜡切片时,切取5片连续切片,其中1片进行HE染色,确认肿瘤细胞的含量。在高灵敏度检测方法中,可考虑使用活检标本。
DNA容易受固定的影响,长时间(1周以上)浸泡在福尔马林中的样本的DNA会被片段化,不能检出突变。活检材料的固定时间一般是24小时,对于穿刺等活检样本,固定时间控制在6~24小时为佳。
4.1.3胸腹水等细胞学样本
用胸腹水中的肿瘤细胞用于基因检测时,必须确认肿瘤细胞,穿刺获得胸腹水样本提交给细胞病理检查之后,剩余液体冷藏/冷冻保存,也可在含有细胞成分的离心沉淀中加入含有蛋白质变性剂的缓冲液(AL缓冲液,Qiagen公司)等室温保存。由于细胞学样本的肿瘤细胞含量较低,因此必须使用高灵敏度检测方法。
4.1.4 血浆样本
循环DNA(circulating free DNA,cfDNA)是存在于血浆中的游离DNA,肿瘤来源的DNA占血浆游离DNA的比例在不同肿瘤及病例中相差悬殊(0.01~93%),从而限制了外周血在肿瘤分子检测时的应用。目前已有多篇文献证实可利用从血浆游离DNA检出突变,但需要使用ARMS法等灵敏度非常高的检测方法。
采集外周血提取血浆游离DNA进行检测,取样时应使用一次性密闭EDTA抗凝真空采血管,采集6~10ml全血,冷藏运输,6小时内分离血浆,提取游离DNA,保存到-80℃冰箱中,并避免反复冻融。如外周血需长时间运输,建议用商品化的游离DNA样本保存管,在常温条件下,cfDNA在全血中可稳定保存7天。
4.2 采样质量的评价
肿瘤细胞是否存在,是开展肿瘤个体化治疗基因检测的重要前提。如果要确认肿瘤细胞存在,与病理诊断医生密切合作是非常必要的。
采样评价内容包括:细胞组成、肿瘤细胞的数量,是否按照要求进行处理与运输。评价方法包括肉眼观察、显微镜下观察和浓度分析等。
肿瘤组织切片等应经病理医师审阅,取一张切片HE染色后显微镜下观察,确保肿瘤细胞存在,并记录肿瘤组织含量,标注肿瘤细胞密集区域。
4.3 样本采集中的防污染
样本采集最好采用一次性材料,不用处理便可直接使用;
制备不同患者病理切片样本时,需更换新刀片,并清除操作器皿上先前样本的残留;
采集中要特别注意防止污染,防止混入操作者的毛发、表皮细胞、痰液等;
如使用玻璃器皿,必须经高压灭菌,以使可能存在的DNase失活;
如提取RNA样品,必须采用RNase抑制剂措施和无RNase材料。
4.4 样本运送和保存
原则上按照《个体化医学检测质量高效指南》要求进行。
实验室应建立详细的样本运送标准操作规程(SOP),对临床医生提供样本采集手册,要求物流人员填写相关运送记录表,确保运送过程中样本的安全性和过程的可控性。
需要转送的样本:用于RNA检测的样本,如果未经稳定化处理,则必须速冻后,放在干冰中运送。
经过适当稳定化处理的样本可在常温下运送,如用于DNA扩增检测的EDTA抗凝全血样本及用于RNA扩增检测的经稳定化处理的样本。
5.分析中质量高效
5.1 实验室设计要求
原则上按照《个体化医学检测质量高效指南》要求进行。在符合国家卫生计生委要求的个体化医学检测实验室和通过审核验收的临床基因扩增检验实验室完成。
5.2 检测方法
5.2.1 Sanger测序法
5.2.1.1技术原理
Sanger测序法即双脱氧链终止法(Chain Termination Method),利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。终止点由反应中相应的双脱氧而定。每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
5.2.1.2技术特点
该方法是DNA序列分析的经典方法,最直接的、可检测已知和未知突变的一种方法。由于该方法可直接读取DNA的序列,因此被认为是基因分型的金标准。
主要优点:测序长度较长,可发现新的变异位点,包括一些新的少见的突变形式及突变的确切类型,如点突变、片段缺失。
局限性:灵敏度不高,突变等位基因需要超过20%才能检出。对样本中肿瘤细胞的含量和比例要求较高,一般要求肿瘤细胞含量不低于50%,如果肿瘤细胞比例低于50%,则假阴性出现的概率会显著增加;不适用于活检或细胞学样本。
5.2.2焦磷酸测序法(Pyrosequencing)
5.2.2.1技术原理
焦磷酸测序技术是由4种酶催化的同一反应体系中的酶级联化学发光反应。当测序引物与模板DNA退火后,在DNA聚合酶、ATP硫酸化酶、荧光素酶和三磷酸腺苷双磷酸酶等4种不同酶的协同作用下,将引物上每一个dNTP聚合时释放的焦磷酸基团(PPi)与一次荧光信号的释放偶联起来,通过检测荧光的释放和强度,达到实时测定DNA序列和定量分析序列变化的目的。
5.2.2.2技术特点
焦磷酸测序技术是一种新型的酶联级联测序技术,其重复性和正确性可与Sanger测序相媲美,而测序速度则大大提高,非常适合对已知的短序列进行重测序分析。
主要优点:检测灵敏度为10%,相对Sanger测序法高,对体细胞突变和甲基化等可实现定量检测;分型正确高效,通量较高,实验设计灵活,可发现新的突变或遗传变异。
局限性:测序长度较短,不能对长片段进行分析。检测灵敏度中等,难以检出低于10%的突变。不适用于活检或细胞学样本。
5.2.3新一代测序 (next generation sequencing,NGS)
5.2.3.1技术原理
NGS又称大规模平行测序(MPS),包含多种可以一次性产生大量数字化基因序列的测序技术,是继Sanger测序的革命性进步,采用平行测序的理念,能够同时对上百万甚至数十亿个DNA分子进行测序,实现了大规模、高通量测序的目标。不同厂家的产品测序原理不同,主要分为边合成边测序(Sequencing by synthesis,SBS)、基于“DNA簇”和“可逆性末端终结(Reversible Terminator)大规模平行测序、 4色荧光标记寡核苷酸的连续连接反应测序和半导体芯片测序。
5.2.3.2技术特点
高通量测序技术不仅可以进行大规模基因组测序,还可用于基因表达分析、非编码小分子RNA的鉴定、转录因子靶基因的筛选和DNA甲基化等相关研究。
主要优点:高通量测序技术有三大优点是传统Sanger测序法所不具备的。第一,它利用芯片进行测序,可以在数百万个点上同时阅读测序。第二,高通量测序技术有定量功能,样品中DNA被测序的次数反映了样品中这种DNA的丰度。第三、利用传统Sanger测序法完成的人类基因组计划总计耗资27亿美元,而现在利用高通量测序技术进行人类基因组测序,测序成本只需1千美金。
局限性:检测灵敏度和测序深度相关,一般来说,NGS在肿瘤体细胞突变检测时,检测灵敏度为10%;已知的与肿瘤相关驱动基因数量有限,疾病表型和基因型的关系还有赖于生物信息的解读,目前NGS应用于肿瘤细胞突变检测的标准化和质量控制尚未形成共识。
5.2.4扩增阻滞突变系统 (ARMS)-PCR法
5.2.4.1技术原理
扩增阻碍突变系统(amplification refractory mutation system, ARMS)是PCR技术应用的发展,也称等位基因特性PCR(allele-specific PCR,AS-PCR)等,用于对已知突变基因进行检测。该法通过设计两个5`端引物,一个与正常DNA互补,一个与突变DNA互补,对于纯合性突变,分别加入这两种引物及3`端引物进行两个平行PCR,只有与突变DNA完互补的引物才可延伸并得到PCR扩增产物。如果错配位于引物的3`端则导致PCR不能延伸。
5.2.4.2技术特点
ARMS-PCR是目前实验室常用的基因突变检测方法。
主要优点:ARMS-PCR法检测灵敏度高,可检测肿瘤细胞中突变比例为1%甚至更低的突变基因。
局限性:只能检测已知的突变类型,不能发现一些新的、未知的突变;如果检测的突变位点或类型较多,则随着引物数目增加出现非特异性结合的概率也相应增加;当检测位点较多时,对DNA样本量的需求增加。
5.2.5 高分辨率熔解曲线(HRM)法
5.2.5.1技术原理
高分辨率熔解曲线(high-resolution melting,HRM)是一种基于PCR新型技术,用于检测基因变异包括未知的基因变异、单核苷酸多态性以及基因甲基化。HRM是基于在加热过程中双链变性为单链的原则。DNA双链体的熔解温度差异反应了基因的变异。双链DNA片段在其特定的温度熔解,熔解的温度由片段的CG含量、序列组成、长度以及一个和多个杂合碱基决定。用DNA嵌合的染料可以看到任何双链片段的熔解峰图,在有荧光嵌合染料的情况下PCR扩增片段,扩增后的产物通过一个快速的可控的加热处理开始熔解。荧光水平在升温的过程中实时监测,染料随着双链DNA的熔解,荧光信号逐渐减少。
5.2.5.2技术特点
由于HRM分析不受碱基突变位点和种类的限制,可用于突变扫描、基因分型、序列匹配、DNA甲基化等方面的研究。
主要优点:检测灵敏度1%,特异性高,重复性好;封闭体系,减少污染的可能性;扩增和检测同时进行,无需PCR后进行处理。
局限性:通过熔解曲线的图不能判断某一特异性的变异体,下游分析中检测需要有测序等补充。
5.2.6 数字PCR(Digital PCR)
5.2.6.1技术原理
数字PCR是一种核酸分子先进定量技术。相较于qPCR,数字PCR能够直接数出DNA分子的个数,对起始样品先进定量。通过将一个样本分成几万到几百万份,分配到不同的反应单元,每个单元包含一个或多个拷贝的目标分子( DNA 模板) ,在每个反应单元中分别对目标分子进行PCR 扩增,扩增结束后对各个反应单元的荧光信号进行统计学分析。数字PCR技术不断发展,Bio-Rad、LIFE Technologies、Fluidigm及RainDance等厂家相继推出技术较为成熟的数字PCR产品。
5.2.6.2技术特点
数字PCR目前的应用包括:稀有等位基因检测、基因表达先进定量、核酸标准品先进定量、二代测序文库先进定量等。
主要优点:灵敏度可达0.001~0.0001%,高特异性,可检测复杂背景下的靶标序列;可高度耐受PCR反应抑制剂;不必依赖对照品或标准品,可对目标拷贝数直接进行正确的鉴定,分析微小的浓度差异;实验数据分析便捷,每个微滴的检测结果以阴性、阳性判读,数据分析自动化;可统计突变率,通过统计分析可得出靶点的突变率。
局限性:数字PCR仪通量较低,目前通常能检测的信号为FAM和HEX。一般单个反应2重反应效果最佳;数字PCR优点是灵敏度高,但是对于DNA浓度大的样本处理就没有优势,而且核酸浓度高时,每个微滴里面包含的拷贝数不符合泊松分布;数字PCR虽然不依赖标准曲线,但是每次反应之间存在差异,短期内不能代替qPCR,也不能代替其他金标准而作为先进方法。QX200等数字PCR仪目前仅用于科研用途,数字PCR走向临床检验还需要一段时间。
5.2.7荧光原位杂交(FISH)
5.2.7.1技术原理
荧光原位杂交(fluorescence in situ hybridization, FISH)是通过荧光标记的DNA探针与细胞核内的DNA靶序列杂交,并在荧光显微镜下观察分析其结果的一种分子细胞遗传学技术。它的基本原理是:如果被检测的染色体或DNA纤维切片上的靶DNA与所用的核酸探针是同源互补的,二者经变性-退火-复性,即可形成靶DNA与核酸探针的杂交体。将核酸探针的某一种核苷酸标记上报告分子如生物素、地高辛,可利用该报告分子与荧光素标记的特异亲和素之间的免疫化学反应,经荧光检测体系在镜下对待测DNA进行定性、定量或相对定位分析。
5.2.7.2技术特点
FISH主要可对基因缺失、基因融合、基因扩增进行检测。
主要优点:可多种荧光标记,显示DNA片段及基因之间的相对位置与方向,空间定位正确;灵敏、特异性好,可同时分析分裂期和间期的多个细胞,并进行定量;可以检测隐匿或微小的染色体畸变及复杂核型。
局限性:FISH检测对操作和判读技术要求较高,诊断医师必须经过严格的FISH操作和结果判读培训,只有经FISH操作经验丰富的医师判定的结果才具有高效性;目前FISH检测的成本昂贵、通量低。
5.2.8免疫组化(IHC)
5.2.8.1技术原理
免疫组化(Immunohistochemistry,IHC)分析利用抗体和抗原之间的结合的高度特异性,借助于组织化学的方法将抗原抗体结合的部位和强度显示出来,以其达到对组织或细胞中的相应抗原进行定性、定位或定量的研究。
5.2.8.2技术特点
IHC作为筛查工具优于FISH,具有经济快捷的优点,尤其适用于大量样本的检测分析。影响IHC结果的因素主要包括抗体的选择、检测前组织的固定,观察者解释方面的差别等。
5.2.9 荧光定量逆转录PCR(Q-RT-PCR)
5.2.9.1技术原理
逆转录PCR(reverse transcription PCR)或者称反转录PCR(reverse transcription-PCR, RT-PCR),是聚合酶链式反应(PCR)的一种广泛应用的变形。由一条RNA单链转录为互补DNA(cDNA)称作“逆转录”,由依赖RNA的DNA聚合酶(逆转录酶)来完成。随后,DNA的另一条链通过脱氧核苷酸引物和依赖DNA的DNA聚合酶完成,随每个循环倍增,即通常的PCR。原先的RNA模板被RNA酶 H降解,留下互补DNA。
5.2.9.2技术特点
荧光定量PCR是检测拷贝数变化的一种快速且经济的技术方法,该方法技术可用于RNA表达水平检测该技术主要优点是检测快速、通量高、灵敏度好,主要不足是对肿瘤组织提取RNA的质量要求较高,在检测基因表达量时判读标准尚未统一。FISH、IHC和Q-RT-PCR三种方法在检测ALK基因重排时的优缺点分析见表1 。
表1:FISH、IHC和Q-RT-PCR检测ALK基因重排的优缺点比较
| IHC | FISH | Q-RT-PCR | |
| 检测对象 | 蛋白 | DNA | RNA |
| 特异性 | + | + | ++++ |
| 发现的融合类型 | 已知、未知 | 已知、未知 | 已知 |
| 通量 | ++ | + | ++++ |
| 费用 | + | ++++ | +++ |
5.2.3 检测方法选择策略
实验室应优选国际和国内“金标准”的检测方法,同类方法中优先选择结果稳定性、重复性好、特异性高的技术,同时也应考虑样本量,检测项目的多少等,综合选择合适的方法。
由于肿瘤组织的异质性,检测的组织样本中混有大量正常组织、石蜡样本提取的DNA质量和数量有限,就检测灵敏度而言,目前ARMS-PCR>焦磷酸测序>Sanger测序。
在检测肿瘤基因突变时,不能一味追求检测方法的灵敏度,灵敏度高的检测方法对整个实验过程中的质控要求更为严格,需防止因污染而产生假阳性。
在肿瘤靶基因表达检测时,由于肿瘤样本的不可再生性,需谨慎选择使用的方法,如选用荧光定量PCR要求提取的总RNA量达1µg以上,如果肿瘤组织过小,提取的RNA量可能达不到检测要求,此时应考虑选用对样本量要求低的检测方法。
5.2.4 检测项目
本指南根据检测靶分子(DNA或RNA)类型及基因表达调控机制类型的不同,将肿瘤个体化治疗检测项目分为基因突变、基因表达、融合基因、基因甲基化检测四个类型。所述检测项目均为目前在肿瘤临床诊疗中,经过严格的循证医学实验,且具有明确的临床应用价值,详见附录1。实验室在开展肿瘤个体化检测项目时,应评估所开展的检测项目的科学性,并遵循实验室的质量管理相关规定。
5.3 DNA提取方法与质量控制
DNA提取之前的病理质量控制非常重要,它决定了最终检测结果的高效性。样本进行检测前均先行常规病理检查和诊断(HE染色),必须经有经验的病理医师确定肿瘤细胞的百分比(肿瘤细胞/整张切片所有细胞,必要时应采取富集肿瘤细胞的方法,如手工刮取或显微切割法。选取以肿瘤细胞为主的、没有明显的坏死、黏液和炎性改变的组织进行检测。
肿瘤细胞比例尚无统一标准,理想情况下,石蜡样本中肿瘤细胞的比例不应低于50%,新鲜样本不低于25%,对于ARMS等敏感性较高的方法,肿瘤细胞的含量和比例可以低一些。具体视所采用的DNA提取方法和突变检测方法的敏感度等而定。
对于新鲜和冻存的手术组织样本,可采用常规的商品化DNA提取试剂盒。对于活检样本,推荐使用能提取石蜡包埋组织微量DNA的试剂盒。对于商品化核酸提取试剂盒,临床实验室在使用前,必须对其核酸提取纯度和效率进行评价。
纯化的靶核酸的完整性可使用凝胶电泳将样本的核酸提取物与核酸标准品比较测定。
核酸提取的产率:可在A260读数测定,DNA可溶于TE溶液中,建议浓度控制在50~100ng/ul,总量20~40 µg或以上。
核酸纯度:可通过提取物A260/A280比率判定,DNA的比值为1.8,RNA的比值为2.0。若DNA比值高于1.8,说明制剂中RNA尚未除尽。RNA、DNA溶液中含有酚和蛋白质将导致比值降低。
5.4 RNA提取方法与质量控制
RNA提取常比较困难,尤其是保存年限较长的肿瘤组织,RNA降解非常严重。造成RNA降解的原因有两个方面:RNA核糖残基的2’和3’位置带有羟基,易被水解;生物体内和外部环境中存在大量RNA酶,并且RNA酶不易失活,高温后仍然能够正确折叠恢复活性。因此从样本的储存、RNA的提取及保存,都需要格外小心,防范RNase对RNA的降解作用。
RNA提取的经典方法是胍盐提取结合酚-氯仿抽提,石蜡样本或活检样本可使用商品化RNA提取试剂盒纯化。细胞或组织的有效匀浆是RNA提取过程中关键的步骤,它能够防止RNA的损失和降解。匀浆的方法应根据细胞或组织的类型来选择。
核酸提取的产率:RNA可溶于无RNase的纯水中,建议浓度控制在100ng/ul以上,总量达30µg以上。
纯化后的RNA应测定OD260/OD280比值,RNA:1.7<OD260/OD280<2.0(<1.7时表明有蛋白质或酚污染;>2.0时表明可能有异硫氰酸残存),进行琼脂糖凝胶电泳观察有无DNA污染和RNA的完整程度。
5.5 试剂的选择、储存及使用注意事项
临床检测试剂必须为CFDA批准的试剂,或具有严格标准操作规程(SOP)的自配试剂(LDT)。
所采用的测定方法特异性好,灵敏度、正确度、精密度符合国家卫生计生委临床检验中心、IFCC、WHO等推荐的方法性能。所用标准品或标准参考物符合国家卫生计生委临床检验中心推荐的标准和要求。
5.6 核酸扩增质量控制
临床PCR检测方法不但要求特异性好、灵敏度高、还要求具有高的可重复性、正确性,尽量降低假阳性、假阴性和非特异性扩增。临床样本扩增时,经常出现假阳性和假阴性,导致检测结果得出错误结论,有时可造成严重后果。
在核酸提取中,应至少带有1份已知弱阳性质控样本。其最后的检测结果,应是核酸提取和扩增检测有效性的综合反映。同时,还应至少带一份已知阴性质控样本,扩增测定的结果可以判断核酸提取过程中是否发生污染。
如靶核酸为DNA,为判断DNA扩增的有效性,可使用1份已制备好的弱阳性靶DNA样本,直接与靶核酸同时扩增检测。如靶核酸为RNA,则除了可用上述弱阳性cDNA判断DNA扩增有效性外,还可用已制备好的弱阳性RNA质控来判断反转录的有效性。另外,须设立阴性对照样品。阴性对照样品检测为阴性时,表明试验全部过程的试剂没有受到核酸污染。在阴性对照样品和阳性对照样品检测结果成立的前提下,才能对检测样品扩增结果进行判定。
5.7 设备维护和校准
实验室仪器的保养与维护是实验室实验技术员的重要组成部分,仪器的保养与维护关系到仪器的适用率、数据的正确率。
仪器设备安装及技术性能验收需由项目负责人、仪器设备使用人、实验室技术人员共同完成,验收内容按采购合同中技术需求部分以服务协议书的要求逐项进行,并且建立仪器设备档案。
仪器设备应定期开展校准计量,未经计量检定、计量检定不合格或未验收的对测试有重要影响的仪器设备不得投入使用。
实验室日常工作中,应重视仪器设备的清洁及维护,做到日维护、周维护、月维护,并及时记录。
5.8 人员培训
检测人员应该有相关的教育背景、相应的工作经验,接受过专业培训。培训主要有内部和外部培训,内部培训包括所使用的试剂方法原理、仪器设备操作维护及校准、质量控制等,外部培训包括国家卫生计生委临床检验中心和国家卫生计生委个体化医学检测培训基地等机构组织开展的各种技术培训。
5.9 方法的性能验证
方法学引进初期需对检测系统测定项目各参数的实验或评估性验证,包括精密度、正确度、线性范围、临床可报告范围、特异性、检测下限、抗干扰能力等。
所有项目的方法验证应形成详细的资料备存,资料需详细描述方法验证的目的、过程、检测结果、分析判断及验证结论。验证的结果及结论须经实验室负责人签字后才能用于临床检测。根据项目的特性,一些方法验证指标不需进行或未进行时,需在报告中书面解释原因。推荐用打印的文稿并在专用文件夹保存。
具体的性能验证方法,如使用的是经CFDA批准的试剂,可参照试剂盒说明书上相应的性能指标部分进行验证,看是否能在其实验室内复现说明书所显示的上述性能指标。如为自制试剂或自建方法,则参考LDT技术指南进行性能评价,建立上述性能指标。以下方法可供参考。
【正确度】
1)参加国家卫生计生委临检中心组织的室间质评,从室间质评统计结果评价实验室检测结果的偏倚或符合情况,从而评价和验证实验室检测结果的正确性。
2)方法比较实验:当引进新方法与原有方法进行比较时,最少样本数为20例,用两种方法同时测定同样的样品。如结果有不符合时,应采用第三种方法或试剂进行确认。
3)检测已知值的标准物质,对于样本来源存在问题的检测方法,可以对已知值的样本稀释成不同浓度再检测,以评估其正确度。
【灵敏度】
1)分析灵敏度:就是确定检测方法的检测下限:将一份已知定值的标准品,用野生型的基因组稀释突变型基因组,设定不同的突变含量,一直稀释到检测下限以下为止,平行检测3管,3管全部检出且其线性∣r∣≥0.98的最低稀释浓度即为该方法的检测下限。(可根据实际情况在最低稀释浓度附近进行10倍以下的稀释)
2)临床灵敏度:主要是验证方法的假阴性率。从三甲医院或专业权威检测机构收集20~50例样本,进行检测分析,其灵敏度应满足临床检测要求,灵敏度=[TB/(TB+FN)]×100(TB=true positive、FN=false negative);有CFDA批文的检测试剂,收集20例阳性样本进行验证;没有CFDA批文的检测试剂或自己实验室研发的LDT试剂,收集50例阳性样本进行验证,并应进行室间比对。
【特异性】
1)特异性的验证主要是确认该方法的假阳性率,特异性=[TN/(TN+FP)]×100(TN=true negative、FN=false positive)。
2)从三甲医院或专业权威检测机构收集20~50例阴性样本,进行检测分析,其特异性应满足临床检测要求。有CFDA批文的检测试剂,收集20例阴性样本进行验证;没有CFDA批文的检测项目或自己研发的项目,收集50例阴性样本进行验证。
6. 分析后质量高效
6.1 检测结果的记录
1)检测结果的报告应正确、清晰、明确、客观和及时,杜绝虚假报告。
2)仪器原始数据要仔细分析,根据质控品判断PCR扩增的有效性,只有当质控品的扩增结果符合项目SOP有关条件时,才可发出报告,否则应重新测定。
3)患者档案及测定结果一并录入“检验管理系统”。报告内容至少应包括:实验室名称、患者姓名、性别、年龄、测定项目、检测方法、检测结果、参考范围、用药建议及样本号、样本类型、检测日期、实验操作者、审核者、报告日期、实验室联系方式等。否则视为无效或虚假报告单。
6.2 失控结果的记录与分析
如发现质控数据违背了控制规则,操作员应填写失控记录或失控报告单,上交实验室主管,由专业主管做出是否发出与失控质控品同批患者样本检测报告的决定。失控信号一旦出现就意味着与失控质控品同批患者样本报告可能作废。此时,首先要尽量查明引起失控的原因,如为假失控,可由实验室指定的资深人员决定、签字后发出报告。如为真失控,最好是纠正原因后,全部样本重新检测,质控品测定合格后再签字发出。
6.3 报告及解释
1)报告的编写需要有严谨有效的流程,以确保检验信息的完整、有效、及时、正确,并保护患者的隐私。检测结果以检测报告单的形式发放,需提供纸质版检测报告,有条件的检测实验室可以电子版的形式发放报告,并建立网络查询系统,送检医生通过登陆网站进行检测结果的查询。
2)检验报告单的应具有患者基本信息、样本情况(采集、送检及检测时间、样本性质及状态等)、检测项目、检测方法、结果、结果的意义、用药建议、检测可能的局限性、检测单位联系方式、检测人员与报告审核人员签字,审核者应当是主管技师以上的工作人员、本专业实验室负责人、中级及以上的病理医师,审核者对检验报告的质量负责。
3)结果解释的责任属于临床实验室,应根据所检测的人群解释结果。临床解释的责任属于临床医生,其应根据检测结果和临床信息向患者解释检测结果。综合临床分子诊断的实验数据和临床信息,为医师和患者描述此结果对疾病诊断的含义,为个体化用药提出建议。
6.4 记录保留
患者和样本信息:接收样本后,在“样本接收记录本”中记录患者和样本信息,对不符合检测要求的样本在“样本拒收登记本”上记录,并及时通知患者。《样本检测申请单》最后保存于扩增区,至少1年以上;其它实验记录保存于各自实验操作区内,至少保存1年以上。《检测过程实验记录》保存于扩增区的专用文件柜内,以备查找。扩增过程中由荧光扩增仪产生的数据文件必须保存在非系统分区的专门文件夹中,定期做备份。
6.5 检测后基因咨询
实验室建立科学的、系统的检验结果解释方案,提供结果的解释意见。报告单上提供咨询服务的方式和途径,如客户服务专线,配置专业的咨询服务人员;方便临床医生和患者随时反馈意见和提出咨询。
6.6 样本(及核酸)保留与处理
肿瘤个体化治疗基因检测报告发出后的样本应尽可能较长期保存。实验室应制定样本储存制度对样本进行保存,建立样本储存的规章制度,做好样本的标识并按规定存放,保存好样本的原始标码,建立配套的样本存放信息管理系统。
样本的处理和相关材料的处理要符合《医疗废物管理条例》、《医疗卫生机构医疗废物管理办法》及国家、地区的相关要求。
6.7 检测与临床数据收集与分析
检测结果收集、整理分析和数据的管理也是保障临床基因检测质量的关键环节。临床的检测结果应定期统计分析,如基因突变的阳性率,如果发现阳性率高于资料报道值,应引起重视,考虑是否存在假阳性情况,如果阳性率偏低,应结合检测方法的局限性考虑假阴性的可能,并进行针对性的改进。
7. 肿瘤个体化治疗检测的质量高效
7.1 标准操作程序
原则上按照《个体化医学检测质量高效指南》的要求进行。肿瘤个体化治疗的检测SOP,应包括试剂准备、样本采集、样本接收与预处理、核酸提取、检测方法、结果分析和报告、仪器操作、实验室安全措施等临床检验的所有环节。SOP的编写应注意通俗易懂、注重细节、清晰明了、图文并茂。实验室工作人员应严格遵循SOP中的步骤要求进行操作,应每年定期根据实验室的运行状况进行SOP审核修订,对于文件的修订、废止、改版或更新,要按照规定的要求,合理且规范进行,并防止无效或作废版本文件使用。
7.2 质控品
检测质控品的建立是为了提高实验室检测结果的高效性和可重复性,一般情况下,针对发现罕见的基因突变、实验运行异常、新批次的试剂与上一批次试剂的比对、储存条件或反应温度发生改变后的样本和试剂、验证整体测试高效性的样本等,都应合理设置质控品。
7.2.1质控品类型
阳性对照:以含有目的片段的DNA(或质粒)作为模板进行扩增,证明PCR试剂是否有效、扩增过程是否正确。但阳性样品扩增效率高,应严格控制其浓度和存放位置,避免其成为潜在的污染源。例如,检测基因突变时,应根据选用的检测方法,选择该方法最低检测限的阳性样本。
阴性对照:以不含有目的片段的阴性样品作为模板进行扩增,用于证明扩增过程中无假阳性现象。
空白对照:以纯水作为模板进行扩增,用于证明扩增过程中无假阳性现象。
PCR抑制物对照:在与阳性对照相同的反应体系中,加入相同数量的待测样品DNA,如果未扩增出目的片段,证明此待测样品DNA中存在PCR抑制物。
空白提取对照:空白提取对照扩增结果为阳性,说明DNA提取试剂或过程中可能受到污染。
阳性提取对照:阳性提取对照扩增结果为阴性,说明提取过程可能有误。如果DNA阳性对照扩增结果为阴性,或者DNA阴性对照和空白对照扩增结果为阳性,则说明PCR试剂或扩增过程存在问题。
基因检测单核苷酸多态性(SNP)时,需要设置多个阳性对照用于检测野生型纯合子基因型、杂合子基因型,突变纯合子基因型。质控应尽可能模拟临床样本,如基质或采样方法与待测样本相同。
7.3 室内质量控制
原则上按照《个体化医学检测质量高效指南》的要求进行。建议按照以下原则设定室内质量控制:
(1)如果只检测1个基因突变,定性测定有一份接近CUT-OFF(2~4倍)的弱阳性和一份阴性质控样本即可。质控品的设置数量随检测样本数的增加而按比例适当增加,例如临床样本数量达到50~60份,则可将阳性和阴性质控样本的数量翻倍。质控品应随机放置在临床样本中间随同临床样本一起同时处理。
(2)当同时检测多个突变基因时,可以根据实验室自身的条件,可只设立能最灵敏地反映检测问题的2个或3个突变基因的阴阳性质控,下次检测改用跟上次不同的基因突变的阴阳性对照,依次类推,循环往复。另外,质控样本在扩增仪中的位置不应持续性的固定在同一个孔,而应在每次扩增检测时,进行相应的顺延,尽可能在一定时间内可以监测到每一孔的扩增有效性。
如果每次质控品的检测结果均成立,说明结果可信,报告可以发出;反之,就要对这种情况出现的原因进行分析。在找不到合理的解释的情况下,须将不符合的基因突变和其相应的对照重新检测。
此外,临床样本中每次检测阳性和阴性结果的出现频率,以及同一基因型或基因突变出现频率或连续出现频率等,均可作为提示实验室“污染”的质控指标。
7.4 室间质量评价
临床检测实验室应参加室间质评(EQA),详细、如实地记录参与EQA的全过程,根据反馈结果了解本实验室的能力、自查存在的问题,及时寻求改进方法,解决问题,完善实验室质量控制体系。
7.5 PCR污染控制
由于肿瘤基因突变检测时必须设置阳性质控品,阳性样本加样时、PCR扩增的产物可能成为PCR实验室的主要污染来源,而PCR的敏感性和效率特别高,所以少量的扩增产物污染样本或反应管即可出现假阳性。尤其是PCR扩增产物和质粒分子很容易造成实验室的污染,导致假阳性现象发生。通过规范实验室设计、规范试剂耗材管理、规范实验室操作和技术处理来控制污染。
常见的PCR污染有样品间交叉污染、PCR试剂的污染、PCR扩增产物污染、气溶胶污染和实验室中克隆质粒的污染。
污染控制:规范实验室设计、规范试剂耗材管理、规范实验室操作和技术处理,如PCR扩增实验中使用dUTP,而不用dTTP。
附录A:常见的检测项目
A.1 基因突变检测项目
A.1.1 EGFR基因突变检测
【基因简介】 EGFR是原癌基因c-erbB1的表达产物,是表皮生长因子受体(HER)家族成员之一。HER家族由EGFR/HER1/erbB1、HER2/neu/erbB2、HER3/erbB3及HER4/erbB4四个分子构成,在细胞的生长、增殖和分化等生理过程中发挥重要的调节作用。
【常见突变】 EGFR的突变主要发生在胞内酪氨酸激酶(TK)区域的前四个外显子上(18~21),目前发现的TK区域突变有30多种。缺失突变主要发生在外显子19上,最常见的是del E746-A750,替代突变最常见的是发生在外显子21上的L858R,复制或插入突变发生在外显子20上。发生在外显子20上的替代突变T790M为耐药突变,研究还发现L858Q、D761Y、T854A等耐药突变。
【EGFR基因突变和EGFR-TKI敏感性】 EGFR-KTI的有效性也因突变类型而不同,外显子19缺失突变的见效率为81%,L858R的见效率为71%,G719X的见效率为56%。吉非替尼初期有效的全部患者,在后期均产生耐药。其中50%患者是在19外显子缺失或L858R点突变等的敏感突变的基础上,又发生了第790位密码子苏氨酸向蛋氨酸的突变(T790M)。研究发现有约1~3%的患者在TKI治疗前即存在T790M,即原发耐药,这种情况下TKI治疗难以有效。
【检测样本类型】 经10%中性福尔马林固定、石蜡包埋的非小细胞肺癌肿瘤组织。
【检测方法】推荐ARMS或Sanger测序法进行EGFR突变检测,可考虑采用新一代测序技术同时进行肺癌驱动基因的检测。
【临床意义】
(1)预测药物疗效:EGFR是HER/ErbB家族信号通路的首要分子,吉非替尼、厄洛替尼等小分子TKI进入细胞内,直接作用于EGFR胞内的激酶区,干扰ATP合成,抑制酪氨酸激酶的活性,阻断激酶的自身磷酸化及底物的磷酸化,有效阻断异常的酪氨酸激酶信号传导,从而阻止配体介导的受体及下游信号通路的激活,阻滞细胞在G1期,促进凋亡,抑制新生血管形成、侵袭和转移,达到治疗的作用。小分子TKI的疗效与EGFR基因突变密切相关,是TKI疗效预测因子。
(2)预后评价:根据是否使用EGFR-TKI对肺癌切除后患者进行预后分析,EGFR敏感性突变并服用TKI的患者至少在单因素分析中有预后良好的趋势。但是,EGFR基因突变与女性、非吸烟者等这些传统的预后良好因子有交叉,只分析基因突变进行预后评价几乎是不可能的。
【用药建议】
(1)吉非替尼、厄洛替尼等小分子酪氨酸激酶抑制剂的疗效与EGFR基因发突变密切相关,特别是当第19外显子缺失、第21外显子突变(L858R)和第18外显子突变(G719X)的患者,使用吉非替尼、厄洛替尼等小分子TKI可获益。
(2)约1~3%未经TKI治疗的NSCLC患者第20外显子存在T790M突变,但经TKI治疗后超过50%后耐药的患者出现T790M突变阳性,导致TKI治疗失败。也有报道认为L747S、D76IY、T854A突变阳性时,患者也会对吉非替尼、厄洛替尼等小分子酪氨酸激酶抑制剂耐药。
【局限性】临床实践表明,并不是所有携有EGFR突变的NSCLC患者都对酪氨酸激酶抑制剂有效,EGFR-TKI的有效性因突变类型而不同,如对外显子19缺失突变的肿瘤患者见效率为81%, L858R的见效率为71%,G719X的见效率为56%,而有些患者发生第20外显子插入突变却对TKI无效。另外,约10%的EGFR野生型NSCLC患者对酪氨酸激酶抑制剂有效,但其机制尚不明确。
A.1.2 KRAS基因突变检测
【基因简介】 哺乳动物基因组中普遍存在三种RAS癌基因家族成员:H-RAS、K-RAS、N-RAS,这三种基因编码的蛋白质大约有90%的氨基酸同源序列,分子量均为21kDa,故称为RASp21蛋白,其在功能上与G蛋白相似,可与二磷酸尿苷(GDP)结合为非活性状态,与三磷酸尿苷(GTP)结合为活性状态,RASp21 蛋白自身具有弱GTPase活性,位于细胞膜内侧参与跨膜信号传递作用。
KRAS基因是RAS基因家族中三种癌基因的一种,位于12号染色体上,含有4个编码外显子和1个5’端非编码外显子,共同编码含189个氨基酸的RAS蛋白。KRAS是表皮生长因子受体功能信号的下游分子,属膜结合型GTP/GDP结合蛋白,通过GTP和GDP的相互转化作用有节制的调节KRAS基因对信号系统的开启和关闭,传递细胞生长分化信号。
【KRAS基因的常见突变】KRAS基因突变发生在肿瘤恶变的早中期,并且原发灶和转移灶的KRAS基因状态基本保持一致。目前研究发现,KRAS基因在膀胱、乳腺、直肠、肾、肝、肺、卵巢、胰腺、胃,还有造血系统等均在一定频率的突变,其中以结直肠癌、胰腺癌和肺癌的发生率比较高,在胰腺癌组织高达90%以上,在肺癌中则以肺腺癌为主,突变率为20~30%,结直肠癌患者突变率为27~43%。当KRAS基因催化活性区突变时,该基因有效活化,不能产生正常的RAS蛋白,导致RAS蛋白不依赖EGFR受体激活而持续活化,造成RAS信号通路的异常活化,影响细胞的生长、增殖和分化,促进细胞的恶性转化,导致细胞增殖失控而癌变。
KRAS基因最常见的突变方式为点突变,90%的KRAS基因突变位于2号外显子的第12和13密码子位点,另有1~4%为第61和146密码子突变。其中结直肠癌中第12密码子(约82%)是最常见的突变位点。一般中国人群样本检测数据G12A高于G12S/C,西方人群相反。
【检测样本类型】 经10%中性福尔马林固定石蜡包埋的结肠癌/肺癌肿瘤组织,或者与原发灶具有同样病理形态的转移组织。
【检测方法】可以采用Sanger测序法,也可采用灵敏度高的ARMS-PCR等。
【临床意义】 西妥昔单抗和帕尼单抗均通过直接抑制EGFR从而发挥抗肿瘤的作用,在结直肠癌和头颈部癌的靶向治疗中都有肯定的效果。西妥昔单抗治疗的有效性受其下游基因KRAS状态的影响,突变型的KRAS无需接受上游EGFR信号即能够自动活化该通路并启动下游信号的转导。因此只有KRAS基因野生型的患者才能从抗EGFR的治疗中获益,而突变型的患者则不能。
【用药建议】KRAS野生型患者使用西妥昔单克隆抗体和帕尼单克隆抗体治疗效果确切,可显著提高患者的生存率和改善生活状态,建议使用;而KRAS的2号外显子的12号密码子和(或)13号密码子或其他密码子任意突变型患者使用西妥昔单抗和帕尼单克隆抗体抗治疗无效,建议不使用该类药物。而在进行结肠癌靶向药物治疗时,应询问所有结肠癌患者的家族史,如果怀疑患者有遗传性非息肉病性结直肠癌(HNPCC)、家族性腺瘤性息肉病(FAP)和轻表型家族性腺瘤性息肉病(AFAP),除非是进行临床试验,否则不应使用贝伐珠单克隆抗体、西妥昔单克隆抗体、帕尼单克隆抗体或伊立替康。
【局限性】临床实践表明,只有50%的野生型KRAS患者对抗EGFR治疗有效,提示EGFR下游信号通路其他分子的激活和变异可能影响了其治疗反应。因此,KRAS基因突变的检测仅用于预测结直肠癌对抗EGFR靶向药物的治疗效果。
A.1.3 BRAF基因突变检测
【基因简介】BRAF基因是1988年由Ikawa等首先在人类尤因肉瘤中发现并克隆确认的一种能转染NIH3T3细胞且有活性的DNA序列。BRAF基因与ARAF、CRAF基因同属RAF家族,命名为鼠类肉瘤滤过性毒菌致癌同源体B1,位于人染色体7q34,长约190kb,编码783个氨基酸的蛋白,相对分子质量为84436,有CR1、CR2和CR3三个保守区。BRAF是Ras-Raf-MEK-ERK信号转导通路重要的转导因子,具有功能的编码区由2510对碱基组成,主要通过有丝蛋白激酶通路中的丝氨酸苏氨酸蛋白激酶来发挥作用,该酶将细胞表面的受体和RAS蛋白通过MEK和ERK与核内的转录因子相连接,启动多种因子参与调控细胞内多种生物学事件,如细胞生长、分化和凋亡。研究表明,在多种人类恶性肿瘤中,如恶性黑色素瘤、结直肠癌、肺癌、甲状腺癌、肝癌及胰腺癌,均存在不同比例的BRAF突变。
【常见突变】BRAF突变主要有两种类型:1.11%位于exon11上的甘氨酸环,如G463、G465、G468的点突变;2.89%的突变发生在exon15上的激活区,其中约92%位于第1799核苷酸上,T突变为A(T1799A以前认为是T1796A),导致其编码的谷氨酸由缬氨酸取代(V600E以前被认为是V599E)。此外,仅不到1%的癌组织同时存在BRAF突变与RAS突变,且在这1%中,BRAF突变几乎均为非V600E突变。以上两种类型的突变均能使BRAF激酶活性及NIH3T3细胞转化能力提高,但以后者更为重要。V600E突变能模拟T598和S601两个位点的磷酸化作用,使BRAF蛋白激活。
【BRAF突变与维罗非尼(vemurafenib)】2011年FDA批准维罗非尼用于治疗晚期(转移性)或不可切除的黑色素瘤,尤其是携有BRAF V600E基因变异肿瘤者。该药的安全性和疗效评估基于一项国际单中心研究。该研究纳入675例BRAF V600E变异的初治晚期黑色素瘤患者,入选者被随机分入维罗非尼组或达卡巴嗪组。结果显示,在维罗非尼组患者未达到中位生存期终点时(77%的患者生存),达卡巴嗪组患者中位生存期为8个月(64%的患者生存)。该药最常见副作用为关节痛、皮疹、脱发、疲乏、恶心和皮肤光敏感。
中国黑色素瘤患者中BREF V600E变异率接近26%,虽然不如白种人(约50%)的变异率高,但仍有可能通过这个药物治疗我国1/4黑色素瘤患者,因此该药物对于黑色素瘤患者的治疗有着十分重要的意义。
【检测样本类型】经10%中性福尔马林固定石蜡包埋的结肠癌肿瘤组织和黑色素瘤组织。
【检测方法】BRAF基因突变的检测方法进可以采用Sanger测序法,也可以使用ARMS-PCR等方法进行检测。
【临床意义】
(1)BRAF是位于KRAS下游级联信号通路上的一个重要蛋白,当BRAF基因发生突变后,其编码生成的蛋白产物无需接受上游信号蛋白的活化便始终处于激活状态,启动下游细胞信号转导途径,引起细胞增殖,从而使EGFR抑制剂西妥昔单克隆抗体和帕尼单克隆抗体等疗效减弱或无效。
(2)BRAF基因可作为患者预后评价的独立性指标,BRAF V600E突变患者呈现预后更差的趋势。
(3)BREF V600E基因突变的黑色素瘤患者对维罗非尼治疗有效。
【用药建议】
(1)对于KRAS基因野生型但同时具有BRAF基因V600E突变的患者,抗EGFR单克隆抗体靶向药物治疗可能无效。(2)回顾性亚组分析显示,无论BRAF基因V600E是否存在突变,一线治疗采用抗EGFR单克隆抗体联合有效的化疗药物都有可能使患者获益。目前有限的研究数据提示,一线治疗后病情进展的患者,如果存在BRAF V600E突变,使用抗EGFR单克隆抗体的疗效欠佳。
(3)50%以上表达BRAFV600突变的晚期黑色素瘤患者在维罗非尼治疗中可获得临床应答。
【局限性】
(1)BRAF基因突变的检测用于预测结直肠癌抗EGFR单克隆抗体靶向药物的治疗效果和预后,必要时必须结合NRAS、KRAS、PI3KCA等基因的突变的检测。
(2)研究发现黑色素瘤患者BRAF V600突变位点外,如携带BRAF L597和K601突变可能对BRAF抑制剂药物维罗非尼药物治疗敏感,但目前还需进一步开展研究来证实这些发现。
A.1.4 C-KIT基因突变检测
【基因简介】C-KIT基因位于人染色体4q11-21,属于原癌基因,其cDNA全长共5230bp,含有21个外显子,编码一个145KD的跨膜酪氨酸激酶受体(tyrosine kinase receptor,RTK)蛋白,命名CD117。第1号外显子编码起始密码子和信号肽,第2-9号密码子编码膜外配体结构域,第10号外显子编码疏水跨膜结构域,第11-20号外显子编码膜内结构域。其中11号外显子编码近膜区段。C-KIT受体属于III型RTK家族,分布于细胞表面,可以用CD117单克隆抗体检测,与血小板衍生生长因子受体(platelet-derived growth factor receptors,PDGFR)有很强的同源性。
【常见突变】多数胃肠道间质瘤(GIST)发生源于C-KIT基因突变,C-KIT突变主要发生在近膜区的外显子11,然后是外膜区的外显子9,酪氨酸区段的外显子13、14、17也可以发生突变。最近数据显示,约8~50%的大肿瘤GIST中可观察到典型的突变,突变频率约35%。在不同GIST中,C-KIT基因突变型式并不有效一样,最常见为第11号外显子突变,导致其编码的近膜结构域的空间结构改变,消弱或丧失对激酶结构域的抑制功能。
【C-KIT基因突变与伊马替尼疗效】甲磺酸伊马替尼(Imatinib mesylate)(商品名:格列卫)是一种口服的酪氨酸激酶抑制剂类药物,能够有效地选择性抑制所有类型的abl酪氨酸激酶活性,包括v-abl、PDGFR和C-KIT蛋白等。伊马替尼于2001年5月在美国上市,同年11月在欧洲上市,并于2002年4月在中国上市。最初被应用于费城染色体阳性的(Ph+)慢性粒细胞白血病(CML)的治疗,之后被批准用于胃肠道间质瘤的治疗,使得GIST治疗进入了分子靶向时代。
一般认为C-KIT/PDGFRA突变类型可以预测伊马替尼的疗效,其中C-KIT 外显子11突变者的疗效最佳;PDGFRA D842V 突变可能对伊马替尼与舒尼替尼原发耐药。舒尼替尼治疗GIST原发C-KIT外显子9突变者和C-KIT野生型者优于C-KIT外显子11突变患者;治疗继发性C-KIT外显子13、14突变患者疗效优于继发C-KIT外显子17、18突变者。
CSCO胃肠间质瘤专家委员会推荐存在以下情况时,应该进行基因学分析:①所有初次诊断的反复和转移性GIST,拟行分子靶向治疗;②原发可切除GIST手术后,中-高度反复风险,拟行伊马替尼辅助治疗;③对疑难病例应进行C-KIT 或PDGFRA突变分析,以明确GIST的诊断;④鉴别NF1型GIST、有效性或不有效性Carney’s 三联征、家族性GIST及儿童GIST;⑤鉴别同时性和异时性多原发GIST。
检测基因突变的位点,至少应包括C-KIT基因的第9、11、13和17号外显子及PDGFRA基因的第12和18号外显子。由于大多数GIST(65~85%)的基因突变发生在C-KIT基因的第11号或第9号外显子,对于经济承受能力有限的患者,在鉴别诊断时,可以优先检测这两个外显子;但是,对于继发耐药的患者,宜增加检测C-KIT基因的13、14、17和18外显子。
【检测样本类型】 经10%中性福尔马林固定石蜡包埋的GIST肿瘤组织。推荐检测的样本类型为治疗前的原发癌肿瘤组织或转移的肿瘤组织。
【检测方法】C-KIT基因突变的检测方法可以采用Sanger测序法、ARMS-PCR等方法检测特定的突变位点。
【临床意义】 (1)辅助诊断和预测疗效:伊马替尼是一种酪氨酸蛋白酶抑制剂,能阻断酪氨酸蛋白激酶KIT受体功能,从而抑制肿瘤的形成。已有研究证实,C-KIT基因突变的位置能影响肿瘤患者对伊马替尼、舒尼替尼等酪氨酸激酶抑制剂的反应。通过检测C-KIT基因的突变状态,协助GIST诊断,也可以进一步的明确诊断CD117阴性的患者,诊断家族性GIST,评价小儿GIST,指导化疗,预测化疗的效果。(2)预后评价:当C-KIT基因第11外显子发生突变后,患者预后较发生于C-KIT基因其他外显子或PDGFRA基因突变的患者或者未检测到C-KIT基因或PDGFRA基因突变的患者预后更差。来源于小肠或结肠的CIST如发生C-KIT基因第9外显子突变,较发生C-KIT基因第11外显子突变者更具有侵袭性。
【用药建议】伊马替尼、苏坦替尼等酪氨酸激酶抑制剂与C-KIT基因发突变密切相关,对发生于外显子9、11、12和17的原发性突变患者,使用伊马替尼、舒尼替尼等酪氨酸激酶抑制剂时,患者可从抗C-TKI靶向药物治疗中获益。当发生位于第13、14、17、18外显子的继发性突变时,则使用伊马替尼、舒尼替尼等酪氨酸激酶抑制剂出现耐药。
【局限性】由于存在肿瘤异质性,或肿瘤组织中混有大量正常组织,或坏死组织过多等,均有可能导致假阴性结果的产生。同时还有部分患者无法提供检测所需的组织样本,或组织样本无法满足检测的基本要求,或患者病情发展及治疗过程中会发生C-KIT基因状态的变化,均可导致治疗的失败。此外,C-KIT基因的突变与伊马替尼、舒尼替尼等酪氨酸激酶抑制剂敏感性之间的关系因人而异。伊马替尼、舒尼替尼等酪氨酸激酶抑制剂在体内的代谢受CYP450 3A4状态的影响,因此即使C-KIT基因突变阳性的患者,伊马替尼、舒尼替尼等酪氨酸激酶抑制剂亦不一定能达到预期临床疗效,需要考虑其他因素的干扰。
A.1.5 PDGFRA基因
【基因简介】PDGFR是分子量为180kD的单链膜糖蛋白,细胞外配体结合区含5个免疫球蛋白样结构域,具保守的半胱氨酸残基,单一跨膜片段;胞内的酪氨酸激酶区断裂处,为亲水氨基酸插入序列。受体分子由α,β两种亚基组成,成熟后的PDGFR以二聚体稳态形式(αα,αβ,ββ)与配体PDGF (platelet-derived growth factor,PDGF)相应异构体(PDGF-AA,AB,BB)结合。结直肠癌组织中PDGFR α和PDGFR β均有表达分布,PDGFR α分布于结直肠正常组织、息肉组织及肿瘤组织上;PDGFR β表达于肿瘤细胞、肿瘤间质细胞和微血管细胞(包括微血管周细胞)上。
【PDGFRA基因的突变】PDGFRA基因突变常见于GIST、胶质母细胞瘤、恶性外周神经鞘等肿瘤,其中GIST中PDGFRA基因突变率在5~10%左右,突变主要发生于PDGFRA基因的近膜端区域(外显子12)和激酶区(外显子14和外显子18),突变频率分别为0.8%和3.9%,其中以外显子18突变为主。PDGFRA常见突变类型见。 PDGFRA基因突变后则通过活化AKT、MAPK及STAT蛋白中STAT1和STAT3发挥作用。也有研究发现活化的A-Raf激酶能调节PLCG1经PDCFR依赖途径的信号转导,也能调节PI3K经PDCFR非依赖的信号转导。
【检测样本类型】经10%中性福尔马林固定石蜡包埋的肿瘤组织。推荐检测的样本类型治疗前的原发癌肿瘤组织或转移的肿瘤组织。
【检测方法】PDGFRA基因突变的检测方法可以采用Sanger测序法、ARMS-PCR等方法检测特定的突变位点。
【临床意义】(1)辅助诊断和预测疗效:伊马替尼是一种酪氨酸蛋白酶抑制剂,能阻断酪氨酸蛋白激酶KIT受体功能,从而抑制肿瘤的形成。已有研究证实,PDGFRA基因突变的位置能影响肿瘤患者对伊马替尼、舒尼替尼等酪氨酸激酶抑制剂的反应。研究表明,PDGFRA基因外显子12和外显子18大部分基因位点突变后使用伊马替尼、舒尼替尼等酪氨酸激酶抑制剂治疗时GIST患者可从中获益。但如外显子18基因位点发生D842V、RD841-842KI或D1842-843IM突变使用伊马替尼、舒尼替尼等酪氨酸激酶抑制剂治疗时GIST患者不能从中获益。(2)预后评价:当PDGFRA基因发生突变后,肿瘤侵袭性较发生于KIT基因突变的患者侵袭性低。
【用药建议】伊马替尼、舒尼替尼等酪氨酿激酶抑制剂与PDGFRA基因发突变密切相关,对发生于外显子12中的Tyr555Cys和Asp56lVal突变及外显子18中的Asp846Tyr等突变患者,使用伊马替尼、舒尼替尼等酪氨酸激酶抑制剂时,患者可从中获益。当位于第18外显子中的D842V、RD841-842KI和DI842-843IM突变时,使用伊马替尼、舒尼替尼等酪氨酸激酶抑制剂则出现耐药。
【局限性】由于存在肿瘤异质性,或肿瘤组织中混有大量正常组织,或坏死组织过多等,无法避免假阴性结果的产生。同时还有部分患者无法提供检测所需的组织样本,或组织样本无法满足进行检测的基本要求,或患者病情发展及治疗过程中会发生PDGFRA基因状态的变化,均可导致治疗的失败。此外,PDGFRA基因突变与伊马替尼、舒尼替尼等酪氨酸激酶抑制剂敏感性之间的关系因人而异。同时伊马替尼、舒尼替尼等酪氨酸激酶抑制剂在体内的代谢受CYP450 3A4状态的影响,即使检测到了PDGFRA基因的突变,使用伊马替尼、舒尼替尼等酪氨酸激酶抑制剂不一定能达到预期临床疗效,需要考虑其他因素的干扰。
A.2 基因表达检测项目
A.2.1 HER2基因表达
【基因简介】原癌基因HER2位于染色体17q21,习惯上称为HER2/neu基因或c-erbB-2基因。编码分子量185kD的跨膜蛋白,因此又被称为p185 HER2,是具有跨膜酪氨酸激酶活性的生长因子受体。HER2受体介导的信号通路是一个复杂的网络系统,包括输入细胞层(配体或生长因子)、信息加工层(受体,SH2蛋白,转录因子)和输出层(细胞生长,分化和转移)。配体介导受体的二聚体是关键,使该信号系统能够传递多种生物信息,而缺乏特异性配体的HER2在整个信号网络中起调节作用。信号转导涉及的主要通路包括:Ras、Raf-Mek-MAPK、PBK、Akt激酶、cAMP(蛋白激酶A)、磷脂酶C-r和src等。HER2通过这些信号转导通路使细胞增殖周期变短,恶性表现增强和抗凋亡。
【HER2基因过表达与乳腺癌等】HER2基因在乳腺癌、膀胱癌、结直肠癌、胃癌和非小细胞肺癌等中均存在表达上调。许多研究资料表明,在20~30%的乳腺癌中存在HER2基因明显扩增或过表达,临床上HER2基因过表达的乳腺癌患者往往表现出生存率低、肿瘤恶性程度增强、进展迅速、易于发生淋巴结转移、化疗缓解期缩短,并对他莫昔芬(Tamoxifen)和很多细胞毒性化疗药耐药等,但对大剂量蒽环类、紫杉类药物疗效好。由于HER2基因位于细胞表面,易被抗体接近,故HER2基因可作为抗肿瘤治疗的一个靶点。
【检测样本类型】经10%中性福尔马林固定石蜡包埋的乳腺癌肿瘤组织。治疗前的原发肿瘤组织或转移的肿瘤组织。
【检测方法】对HER2基因表达的检测方法可以采用FISH、IHC、扩增显色原位杂交(CISH)等,目前也有实验室使用荧光定量PCR等方法进行检测,但该方法用于检测HER2基因的表达尚未得到承认。一般来说,实验室首先采用IHC方法进行HER2蛋白检测,如果检测结果为2+,则进行原位杂交(FISH)方法进行HER2基因检测确认。
【结果判读】
免疫组织化学(IHC)检测:
1)3+,HER2表达阳性;
2)1+或阴性,表达阴性;
3)2+时则需要进行FISH检测。
FISH检测:
1)HER2与CEP17信号数比值:≥2.0为阳性,有HER-2/neu基因扩增;
2)HER2与CEP17信号数比值:<2.0时,
若HER2拷贝数≥6.0 为阳性,有HER-2/neu基因扩增;
若 HER2拷贝数<4.0 为阴性,无HER-2/neu基因扩增;
若 HER2拷贝数≥4.0,但<6.0时为不确定,不能确定HER-2/neu基因状态。
3)若众多信号HER2信号连接成簇时可不计算,即视为基因扩增。
【临床意义】正确分析HER2基因扩增状态是乳腺癌患者预后判断及制订有效治疗方案的先决条件,对乳腺癌的诊疗具有重要的指导作用。
(1)预后评价:研究显示,HER2基因的过表达除了与肿瘤的发生发展相关外,还是一个重要的临床预后指标,主要表现为HER2基因扩增的乳腺癌浸润性强、无进展生存期(progress free survival、PFS)短、预后差。而且这部分患者就诊时的肿瘤负荷更大,淋巴结转移的几率更高,激素受体阴性的比例更高、组织学分级更差、肿瘤的增殖指数更高、反复风险更高。但没有证据显示HER2基因扩增与导管原位癌( ductal carcinoma in situ,DCIS)的预后相关。
(2)内分泌药物疗效预测:研究显示,相对于无HER2基因扩增的乳腺癌患者而言,HER2基因扩增的患者应用他莫昔芬治疗后的死亡风险明显增高,因此这类乳腺癌患者可能不适合选择他莫昔芬作为内分泌治疗,而且HER2基因扩增的乳腺癌患者对CMF化疗方案的反应性降低,宜采用高剂量的蒽环类药物方案。
(3)靶向药物疗效预测:大量临床研究数据提示,使用曲妥珠单克隆抗体等治疗乳腺癌时,无论是与常规化疗联合用于乳腺癌患者的辅助治疗,还是用于辅助治疗后的维持治疗,及用于晚期乳癌患者的单药或联合治疗,都能肯定改善HER2基因扩增或蛋白过表达患者的生存,使乳腺癌患者获益。
即使在使用曲妥珠单抗治疗过程中出现疾病进展而需要进一步治疗的乳腺癌患者,继续使用曲妥珠单抗治疗仍然有效。而对于HER2基因低度扩增或不扩增的乳腺癌患者,使用曲妥珠单抗疗效不佳。
【用药建议】曲妥珠单抗及拉帕替尼等酪氨酸激酶抑制剂等乳腺癌靶向药物治疗乳腺癌的疗效与HER2基因表达状态密切相关,当HER2基因扩增时,使用曲曲妥珠单抗和拉帕替尼等酪氨酸激酶抑制剂时,患者可从抗HER2靶向药物治疗中获益。但如果发生PI3KCA基因突变、PTEN基因失活及HER2基因某些位点发生突变时,则会对曲妥珠单抗和拉帕替尼等酪氨酸激酶抑制剂耐药。
【局限性】由于方法学本身的局限性,IHC和FISH得出的均有不确定结果,无法对HER2基因状态做出明确判断。即使经IHC或FISH判断为HER2基因过表达的患者也未必一定能从靶向治疗中获益,导致这种现象的原因可能是检测体系本身所造成的假阳性,也可能是HER2基因的信号通路中还存在其他异常的位点。其次肿瘤异质性的存在导致IHC和FISH均无法避免假阴性结果的产生。还有部分患者无法提供检测所需的组织样本,或组织样本无法满足进行检测的基本要求,或患者病情发展及治疗过程中会发生HER2基因状态的变化,而IHC和FISH均无法对患者的HER2基因状态进行动态、实时的监测。
A.3融合基因检测项目
A.3.1 EML4-ALK融合基因检测项目
【基因简介】ALK,即人类间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK),于1994年首先发现于间变性大细胞淋巴瘤AMS3细胞株中,是由1620个氨基酸组成的跨膜蛋白,属于胰岛素受体家族。EML4是人类棘皮动物微管相关蛋白样4(echinoderm microtubule-associated protein- like 4,EML4),属于棘皮动物微管相关蛋白样蛋白家族,由N末端碱基区、疏水的棘皮动物微管相关蛋白区(hydrophobic echinoderm microtubule-associated protein-like protein,HELP)及WD重复区三部分构成。该融合基因定位于2号染色体的短臂上(2p21和2p23),其5’端为EML4的片段,3’端为ALK的片段,由倒置后的EML4基因片段与残余的ALK片段连接。该融合基因拥有EML4基因中的BASIC区域,疏水的棘皮动物微管相关蛋白区及部分WD重复区(后两部分在部分亚型中缺失)和ALK基因中的Kinase功能区。EML4-ALK 的信号转导通路为PI3-K/Akt、STAT3/5、Ras-MEK 和PLC-γ/PIP2等,这些通路与细胞存活、增殖和迁移密切相关。
【EML4-ALK融合与克唑替尼】克唑替尼是一种酪氨酸激酶受体抑制剂,靶向分子包括ALK、肝细胞生长因子受体(HGFR,c-Met)和ROS1,于2011年获得美国食品和药品管理局(FDA)批准用于治疗间变型淋巴瘤激酶(ALK)基因重排的非小细胞肺癌(NSCLC)。EML4-ALK基因融合可促使ALK基因引起致癌融合蛋白的表达。ALK融合蛋白形成可引起基因表达和信号的激活和失调,进而促使表达这些蛋白的肿瘤细胞增殖和存活。克唑替尼在肿瘤细胞株中对ALK和c-Met在细胞水平检测的磷酸化具有浓度依赖性抑制作用,对表达EML4-ALK或NPM-ALK融合蛋白或 c-Met的异种移植荷瘤小鼠具有抗肿瘤活性在NSCLC患者中,ALK重排的阳性率大约为3~5%,在腺癌、从未吸烟或少量吸烟的患者中EML4-ALK融合的几率高。
【检测样本类型】经10%中性福尔马林固定、石蜡包埋的非小细胞肺癌肿瘤组织。推荐检测的样本为治疗前的原发癌肿瘤组织或转移的肿瘤组织
【检测方法】EMIA-ALK融合基因的检测方法有FISH、IHC、荧光定量PCR等,推荐的检测方法为FISH。
【临床意义】(1)预测药物疗效:EMLA-ALK融合基因阳性的NSCLC患者接受以铂类为基础的化疗,其见效率、疾病进展时间和总生存期与EGFR突变阳性NSCLC患者相似。相反,EML4-ALK融合基因阳性患者不能从EGFR-TKI的基础治疗中受益,表现为原发耐药,治疗结果与无EGFR基因突变的患者相似。而针对EML4-ALK融合基因阳性的患者,使用克唑替尼等针对ALK基因的小分子抑制剂可以获得良好的临床治疗效果。因此在使用针对ALK基因的小分子抑制剂前,需进行EML4-ALK融合基因突变的检测。
【用药建议】针对ALK基因的小分子抑制剂疗效与EML4-ALK融合基因密切相关,当存在EML4-ALK融合基因时,可以考虑使用针对ALK基因的小分子抑制剂治疗如克唑替尼,患者可以从中获益,而不应给予吉非替尼、厄洛替尼等EGFR-TKI类药物,患者不会从中获益。
【局限性】由于EML4-ALK融合基因检测方法FISH、IHC和RT-PCR检测的灵敏度不足及检测样本受正常组织干扰等因素的影响,容易造成检测结果的假阴性。同时EML4-ALK融合基因各种亚型患者在接受克唑替尼治疗时是否存在疗效差异尚不明确,也有待进一步研究。因此,使用克唑替尼治疗EML4-ALK融合基因阳性的NSCLC患者时,需要定期监测疗效。
A.4 基因甲基化检测项目
A.4.1 MGMT基因甲基化检测
【基因简介】人MGMT基因稳定地存在于所有正常组织细胞内,其编码的MGMT蛋白以分布在人体肝脏的活性最高,其次为淋巴结和肠道,骨髓细胞中的活性最低。MGMT蛋白在不需要任何辅助因子或其他蛋白质的条件下,可以催化DNA分子中鸟嘌呤O6位上的烷基转移至MGMT本身第145位的半胱氨酸残基上,鸟嘌呤损伤修复,DNA的结构和功能得以恢复。同时,这种催化作用是一种不可逆的反应,MGMT作用后由于获得烷基而失活,因此这种酶又是一种自杀蛋白。正常情况下,细胞内MGMT具有解除烷化剂对细胞的致癌作用和消除烷化类药物对于细胞毒性杀伤作用的双重生物学功能,而细胞对DNA鸟嘌呤O6位上烷基化修复能力的大小通常取决于MGMT在细胞内的含量和合成的速率。
【MGMT基因启动子甲基化】影响机体MGMT含量和活性的因素有很多,环境因素、机体器官状态和基因状态等。其中基因调节是影响MGMT蛋白含量和活性的主要因素,MGMT基因启动子甲基化是MGMT最常见的异常,多发生于MGMT启动子CpG岛,导致该基因转录停止,表达减少。许多肿瘤如脑胶质瘤、结直肠癌、肺癌、乳腺癌中存在MGMT基因甲基化并均可观察到MGMT启动子异常甲基化,启动子甲基化与19号染色体长臂丢失或19号染色体长臂与1号染色体短臂杂合丢失有关。如p53基因突变后能导致肿瘤细胞MGMT表达减少,活性降低。许多机体环境因素也影响MGMT蛋白的含量和活性,如乙基亚硝基脲、吸烟等可以使肝细胞中的MGMT蛋白表达量和活性明显增加。MGMT启动子发生甲基化的患者才可以从替莫唑胺治疗中受益。
【结果解释】正常人群基因组中MGMT基因启动子CpG位点为非甲基化状态,如果检测到基因组中存在MGMT基因启动子CpG位点的甲基化,提示MGMT基因编码MGMT蛋白的功能下降或MGMT活性降低。
【检测样本类型】经10%中性福尔马林固定石蜡包埋的脑胶质瘤肿瘤组织或者病理确证的活检组织。推荐检测的样本类型为治疗前的原发癌肿瘤组织。
【检测方法】MGMT基因启动子甲基化的检测方法建议采用MSP (methyl-specific polymerase chain reaction,甲基化特异的PCR)、甲基化特异性焦磷酸测序、HRM等方法。
【临床意义】(1)疗效预测: MGMT启动子发生甲基化的患者明显比未发生甲基化的患者使用烷化剂的疗效好,其总体生存率和无进展生存率更高。MGMT启动子区甲基化对胶质瘤一线化疗药物TMZ治疗胶质瘤的化疗疗效具有预测价值,且是独立的预后较好的指示指标。MGMT启动子未甲基化者从TMZ常规治疗方案中获益较小,应对这类患者采用更有效的有助于克服耐药的其他化疗方案。(2)预后评价:40%脑胶质瘤患者有MGMT启动子甲基化,甲基化程度越高,预后越差,其对肿瘤的预后和生存期的预示作用较肿瘤的分级、临床、年龄等其他特征更有效。
【用药建议】TMZ等烷化剂疗效与MGMT启动子区域甲基化状态密切相关,对发生了MGMT启动子区域甲基化的患者,且发生甲基化比例越高的患者,使用TMZ烷化剂治疗时,患者可从中获益。如未发生MGMT启动子区域甲基化的患者,使用TMZ烷化剂治疗则出现耐药。
【局限性】由于脑部肿瘤的特殊性,限制了肿瘤组织的来源,同时肿瘤组织的异质性、大量坏死组织或大量正常组织的存在,干扰了检测结果,导致检测结果的假阴性。同时,MGMT启动子区域甲基化程度与TMZ烷化剂的疗效之间的关系尚未阐明,MGMT基因表达水平也影响了TMZ烷化剂的疗效,且TMZ疗效也受其他因素的影响,因此尚不能根据MGMT启动子甲基化状态判断TMZ的疗效,仅仅是根据是否存在甲基化对TMZ的疗效进行预测。
参考文献:
[1] Molecular Methods for Clinical Genetics and Oncology Testing; Approved Guideline—Third Edition MM01-A3
[2] Collection, Transport, Preparation, and Storage of Specimens for Molecular Methods; Approved GuidelineMM13-A
[3] Quality Management for Molecular Genetic Testing; Approved Guideline MM20-A
[4] Comparison of immunohistochemistry with fluorescence in situ hybridization in determining the human epidermal growth factor receptor 2 status of breast cancer specimens: a multicenter study of 3 149 Chinese patients. Chin Med J 2014;127 (2): 246-253
[5] 李艳,李金明. 《个体化医疗中的临床分子诊断》 人民卫生出版社 2013年8月
[6] NCCN临床实践指南:非小细胞肺癌(2015.V1)
[7] NCCN临床实践指南:结直肠癌(2015.V1)
[8] NCCN临床实践指南:乳腺癌(2015.V1)
- 【佳学基因检测】前列腺癌的基因突变检测的部分位点和内容(新增)...
- 【佳学基因检测】乳腺癌基因检测报告怎么看...
- 【佳学基因检测】乳腺癌28项基因检测价格...
- 【佳学基因检测】乳腺癌遗传因素大吗?...
- 【佳学基因检测】低遗传力性状有哪些...
- 【佳学基因检测】乳腺癌基因测定...
- 【佳学基因检测】乳腺癌基因检测正确率达到多少...
- 【佳学基因检测】乳腺癌基因检测结果分析...
- 【佳学基因检测】乳腺癌基因检测有哪些项目...
- 【佳学基因检测】乳腺癌基因检测项目名称...
- 【佳学基因检测】21基因多少不化疗...
- 【佳学基因检测】乳腺癌哪种病理最好...
- 【佳学基因检测】佳学基因解码基因检测...
- 【佳学基因检测】基因是什么...
- 【佳学基因检测】乳腺癌遗传测试方法...
- 【佳学基因检测】乳腺癌遗传测试怎么做...
- 【佳学基因检测】乳腺癌遗传检测...
- 【佳学基因检测】乳腺癌遗传性检测...
- 【佳学基因检测】乳腺癌遗传筛查...
- 【佳学基因检测】乳腺癌遗传基因怎么检测...
- 【佳学基因检测】乳腺癌遗传基因检测查哪些项目...
- 【佳学基因检测】乳腺癌遗传基因筛查...
- 【佳学基因检测】肿瘤基因检测价格表...
- 【佳学基因检测】基因检测的费用大概多少钱...
- 【佳学基因检测】HER2阳性是最轻的乳腺癌吗...
- 【佳学基因检测】不化疗和化疗哪个寿命长...
- 【佳学基因检测】乳腺癌基因检测报告怎么看...
- 【佳学基因检测】乳腺癌基因检测的费用大概多少钱...
- 【佳学基因检测】乳腺癌基因检测有必要吗...
- 【佳学基因检测】乳腺癌基因检测的作用与意义...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
