












【佳学基因检测】视锥视杆细胞营养不良基因检测(Cone rod dystrophies)
为什么要做视锥视杆细胞基因检测?
锥体-杆体视网膜变性(视锥视杆细胞营养不良)(发病率 1/4万)是一组属于色素性视网膜病变的继发性视网膜变性。视锥视杆细胞营养不良的特征是眼底检查可见视网膜色素沉着,主要局限于黄斑区。与典型的视网膜色素变性(RP,也称为杆体-锥体视网膜变性RCDs)不同,RCDs是由于杆体光受体的原发性丢失,随后锥体光受体继发性丢失引起的;而视锥视杆细胞营养不良反映了相反的事件顺序。CRD的特征是锥体的原发性受损,有时是锥体和杆体同时丢失,这解释了视锥视杆细胞营养不良的主要症状:视力下降、色觉缺陷、闪光和中央视野灵敏度下降,后来逐渐出现外周视野和暗 adaptation 丧失。视锥视杆细胞营养不良的临床过程通常比RCDs更严重和快速,导致更早的法律失明和残疾。但是,在末期阶段,视锥视杆细胞营养不良与RCDs没有区别。视锥视杆细胞营养不良贼常见的是非综合征性的,但也可以是几种综合征的一部分,如巴德-比德尔综合征和小脑性共济失调7型(SCA7)。非综合征性视锥视杆细胞营养不良在遗传学上异质性很大(到目前为止已经克隆了十个基因和确定了三个位点)。与视锥视杆细胞营养不良发病机制相关的四个主要致病基因是ABCA4(引起斯塔格德氏病,也占了30-60%的视锥视杆细胞营养不良),CRX和GUCY2D(这两个基因导致了许多报告的视锥视杆细胞营养不良的自动主性遗传形式),以及RPGR(引起约2/3的X连锁RP,也占不确定比例的X连锁视锥视杆细胞营养不良)。高度致病性突变在通常引起RP或黄斑变性的基因中也可能导致视锥视杆细胞营养不良的可能性很大。视锥视杆细胞营养不良的诊断依赖于病史、眼底检查和视网膜电图。 一些基因可以进行分子诊断,遗传咨询总是被建议的。目前没有可以阻止疾病进展或恢复视力的治疗方法,视觉预后不佳。治疗的目标是减慢退行性过程、治疗并发症,帮助患者应对致盲的社会和心理影响。
疾病名称
锥体-杆体视网膜变性(视锥视杆细胞营养不良), Cone rod dystrophies
什么是视锥视杆细胞营养不良及其诊断
视锥视杆细胞营养不良是属于色素性视网膜病变组的继发性视网膜变性。
视锥视杆细胞营养不良的功能性体征和症状
- 贼早出现的症状是视力下降
- 早期也出现光恐惧
- 频繁的失色觉
- 暗 adaptation 丧失出现在较晚
患者的视野变化
- 贼早出现中央暗点,影响流畅阅读
- 后来逐渐出现散在的外周视野损害
- 与RP相比,严重的视力损失发生得更早
眼底
早期阶段,正常的黄斑或细小的黄斑病变和视神经盘苍白可被视为少有的体征
骨刺样的色素沉着,常见于黄斑区
视网膜血管变细
视神经盘蜡样苍白
不同程度的视网膜萎缩
视网膜电图(ERG)
30Hz闪光刺激下隐含时间(a波和b波峰值之间)移位,以及a波和b波单闪光峰延迟,是振幅降低前的早期体征
a波和b波幅度显著下降
光敏响应(锥体)比暗敏响应(杆体)受损更严重
流行病学
视锥视杆细胞营养不良的发病率估计为1/4万(因此,视锥视杆细胞营养不良比RP少10倍)[1]。
临床描述
非综合征性锥体-杆体视网膜变性
视锥视杆细胞营养不良起初表现为黄斑病变或以黄斑参与为主的弥漫性视网膜病变。与杆体-锥体视网膜变性(RCDs,典型的视网膜色素变性)的症状不同,RCDs是由杆体优先受损导致的夜盲和外周视野损失,视锥视杆细胞营养不良的临床体征反映了锥体的主要参与,这导致视力下降和中央视野敏感度损失。这符合CRD实体的原始描述,其中锥体损失发生在杆体变性之前。 然而,在某些情况下,弥漫性视网膜病变同时影响锥体和杆体,导致夜盲和视力下降。这些病例也可以视为视锥视杆细胞营养不良,尽管它们与其他实体重叠(见鉴别诊断)。 总的来说,视锥视杆细胞营养不良比RCDs严重,因为患者自主活动能力的丧失发生得更早。 将CRD的病程描述为两个阶段是方便的。
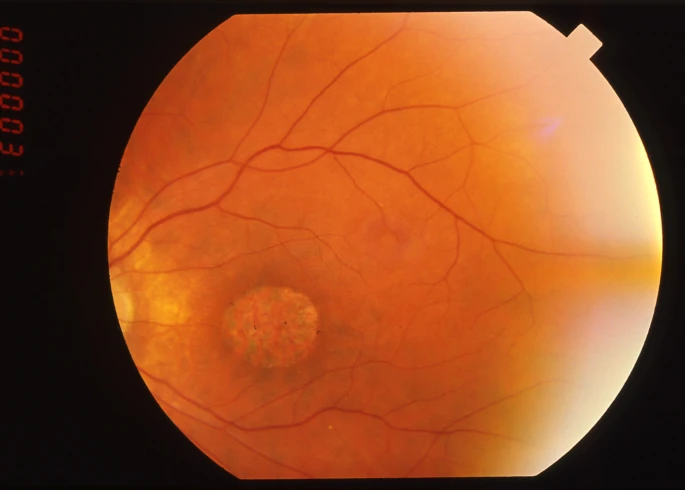
在先进阶段,主要症状是视力下降,通常在十年内的先进十年被发现,而且不能通过配镜显著改善。患者经常有显着的凝视倾斜,以投射图像到视网膜较不受损害的视网膜旁中心区。 伴随这一症状,有强烈的光恐惧和不同程度的失色觉。相比之下,夜盲无被患者提及或在存在时也从未像视力下降那样突出。 视野检查显示中央暗点,而外围保留。因此,患者移动没有困难。 眼底检查显示黄斑区域色素沉着和不同程度的视网膜萎缩(图1)。视网膜血管通常正常或中度变细。视神经盘在早期阶段经常苍白,特别是在颞侧,这与黄斑纤维束相符。 在这个阶段,需要区分视锥视杆细胞营养不良和斯塔格德氏病等黄斑变性、锥体变性和其他少见的黄斑状况。 额外的检查有助于诊断。 首先,荧光素血管造影和眼底自发荧光显示外周视网膜也受累,但荧光不均匀性程度较黄斑轻。 其次,视网膜电图(ERG)显示锥体响应的隐含时间移位,之后是锥体和杆体响应的减少。 锥体反应受损更严重。

在第二阶段,夜盲变得更加明显,外周视野损失继续加重。因此,患者独立活动有困难。此外,视力持续下降到无法再进行阅读的水平。眼肌阵挛经常存在。在这个阶段,患者在法律上已属盲人(视力<1/20),尽管大部分外周视野仍保持。
综合征性锥体-杆体视网膜变性
有一些综合征的视网膜退行性变表现为视锥视杆细胞营养不良而不是典型的RP。
• 巴德-比德尔综合征(BBS)是一种常染色体隐性疾病,发病率介于1/13500至1/60000之间。它将视网膜萎缩与骶后多指症、肥胖、性腺发育不良、精神发育迟缓或轻度精神运动发育迟缓和可能导致肾功能衰竭的肾病变联系在一起。视网膜萎缩经典地被描述为RCD,但据报道有许多变体具有突出的黄斑参与(图2),表示CRD [2]。事实上,BBS患者属于弥漫性CRD。根据我们的经验,他们的黄斑总是参与,表现为视力下降、光恐惧及荧光素血管造影中黄斑区超荧光。视网膜萎缩的诊断通常在十年内建立,法律盲目在20岁前达到,但疾病有中等形式。当临床表现不完整时,诊断可能有困难。在这种情况下,CRD的存在是一个重要的体征。到目前为止,已报道12个BBS基因编码参与纤毛结构的蛋白。
• 小脑性共济失调7型是一种由积聚的polyglutamine在ataxin蛋白中的扩增引起的自动主性小脑共济失调性病变。视网膜病变通常从颗粒状黄斑开始,逐渐扩散到整个视网膜,而黄斑变为萎缩(图3)[3]。起初,疾病通常表现为孤立的视网膜萎缩;特征性的黄斑参与和视功能损害的重要性在此前视力良好的患者中应该引起神经系统检查。
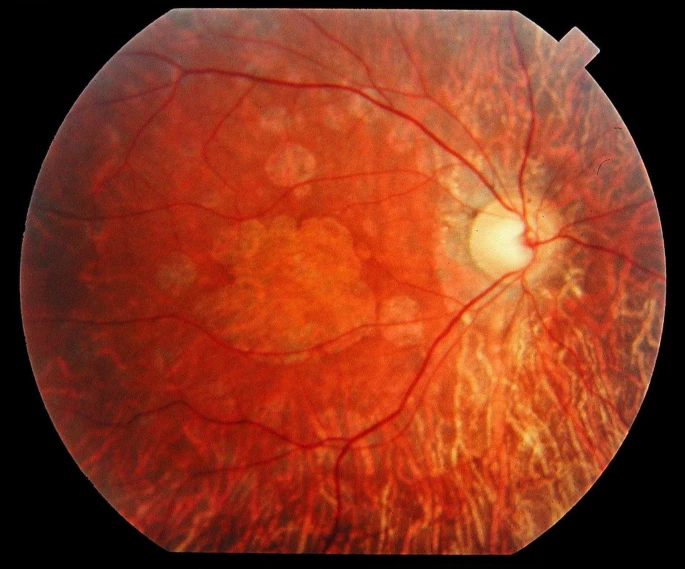
图3:小脑性共济失调7型(SCA7)引起的锥体-杆体视网膜变性34岁患者的眼底照片。注意黄斑区和中周围都发生了萎缩。
• 外胚层疾病。CRD有时见于:
○ 釉质发育不良。这指的是几种牙釉质异常的条件。一种与CRD和畸形牙齿相关的常染色体隐性釉质发育不良(OMIM # 217080)
○ 幼年型黄斑变性伴发hypotrichosis。这是一种由佳学基因检测进行检测与分析常染色体隐性脱发伴发黄斑变性。通常视网膜损害限于黄斑,但在少数情况下也有报道为CRD。
○ 畸形综合征。已在软骨干骺发育不良和裂唇中报道视锥视杆细胞营养不良。
○ 代谢功能紊乱。已报道CRD发生在几种代谢性疾病中(硫胺响应性巨母细胞贫血;已报道一例线粒体突变(T8993G)病例[13])。此外,植酸增多的婴儿型Refsum病与色素沉着视网膜病变相关,特征性表现为明显的黄斑参与。在Alport综合征(耳聋、进行性肾炎)中,眼底表现为类似晶体的白色斑点散布于黄斑周围,而不是真正的色素沉着视网膜病变。
非综合征性视锥视杆细胞营养不良的病因学
与典型RP一样,非综合征性视锥视杆细胞营养不良在遗传学上也异质性很大。已报道三种门德尔遗传类型。如今,已知13个非综合征性视锥视杆细胞营养不良的致病基因(10个已克隆,3个已定位)。这些基因可以分为几个类别。
先进类主要包括视锥视杆细胞营养不良病例中的致病基因。其中占主导地位的是编码家族转录因子CRX的基因,它控制光感受器细胞的分化和生存。大多数CRX突变导致自动主性CRD,其发病率估计占主导性视锥视杆细胞营养不良的5%至10% [14,15]。该疾病的严重程度差异很大,有些为轻度,有些为非常严重。事实上,在这一范围的末端,已有几篇报道称CRX突变可导致主导性Leber先天性青光眼(LCA)[16,17],后者通常为隐性,也可导致少数RP。仅在视锥视杆细胞营养不良中发现了另外两个基因。这些是RIM1,报道于一个自动主性CRD家系中[18],以及HRG4,报道于一个继承方式不明的家系中[19]。有趣的是,这两种编码蛋白都参与光感受器突触传递。
第二类主要包括在黄斑变性中发现的基因。如今,它主要包含一个基因ABCA4,这涉及视网膜素代谢并引起斯塔格德氏病。 ABCA4基因突变负责30%至60%的自动体隐性视锥视杆细胞营养不良病例。在某些情况下,疾病起初表现为斯塔格德氏病,很快扩展至外周;在其他情况下,疾病起初表现为以黄斑参与为主的弥漫性视网膜病变。已证明与视锥视杆细胞营养不良相关的ABCA4突变为截短突变,常在两个等位基因上同时存在,而氨基酸改变突变更经常见于斯塔格德氏病中。这表明更有害的截短突变与CRD等更严重的病况相关[26]。属于这一类别,在一个自动主性CRD家系中已描述GUCA1A的突变,而所有其他GUCA1A突变都导致锥体变性。 GUCA1A基因编码一种激活鸟苷酸循环酶(GC)的蛋白。GC本身有时也参与视锥视杆细胞营养不良(见下文)。
第三类主要包括在RP病例中发现的两个基因。一个编码外节蛋白peripherin/RDS,通常参与主导性RP。众所周知,RDS突变与主导性黄斑变性或主导性CRD存在家族内和家族间的表型变异。与自动体隐性视锥视杆细胞营养不良相比,RDS基因突变引起的视锥视杆细胞营养不良相对温和,因为患者的自主活动能力在成年早期得到保持。另一个基因编码RPGR(参与视蛋白转运,特别是锥体视蛋白)。RPGR是X连锁RP的主要致病基因,但它也解释了一些X连锁CRD,其位点被称为COD1或CORDX1,以及一些锥体变性。与RPs一样,RPGR基因突变引起的视锥视杆细胞营养不良严重,并且在生命早期被诊断出来。除了RDS和RPGR,CACNA1F基因(其突变导致X连锁 stationary night blindness)在一个之前被映射为CORDX3(或COD4)的芬兰CRD家系中被发现突变。
第四类包括在LCA中发现的基因。如今,已报道3个RPGRIP1基因突变的CRD家系31和AIPL1 32。这些基因通常参与LCA的发病机制。还报道了几个GUCY2D基因突变的CRD家系,GUCY2D是LCA的主要致病基因。与LCA患者相比,GUCY2D突变的CRD患者表现出主导性,突变仅限于编码鸟苷酸循环酶二聚体化结构域的外显子13。
还有一些基因有待克隆的位点。 对于自动主性视锥视杆细胞营养不良,在一个与CRD和精神发育迟缓相关的丹麦家族中发现了CORD1基因(未确定定位)。 CORD4已在一个与神经纤维瘤病相关的加拿大CRD家系中定位。 对于自动体隐性视锥视杆细胞营养不良,有定位在一个巴基斯坦家族中的CORD8和一个巴西家族中的CORD9。 对于X连锁视锥视杆细胞营养不良,CORDX2已被定位到Xq27。
总的来看,大多数视锥视杆细胞营养不良致病基因似乎也参与其他类型的视网膜变性,包括RP、黄斑变性和锥体变性,这将视锥视杆细胞营养不良置于广泛的视网膜变性谱系的中心。 因此,任何导致视网膜变性的基因都可能潜在地参与CRD的发病机制,挑战在于理解基本机制。 很明显,视网膜变性基因的有害突变可以导致非常严重的疾病,因此导致视锥视杆细胞营养不良。 然而,为什么在某些家族中,某些成员有黄斑变性或RP,而其他成员(具有相同突变)有CRD,原因还不清楚。 同样,为什么某个基因的某些突变导致CRD而其他突变引起RP,对于几个基因来说仍未解决。
诊断方法
临床诊断依据视力早期下降和光恐惧、眼底病变、以锥体受损为主的低电压ERG踪迹以及这些体征的逐渐加重。 全视野ERG是关键的测试,特别是在患者无症状且早期眼底正常的情况下。 重要的是通过在贼初建立诊断后1-2年重复检查来确定诊断。 多黄点ERG可用于正确地监测中央视网膜的功能。
目前,由于疾病的巨大遗传异质性,系统的分子测试还没有成为例行的。 然而,快速和大规模的突变筛查技术正在发展,几个实验室正在对贼常涉及的基因(包括ABCA4、CRX、GUC1A)进行突变筛查; 测试单个患者DNA中的几十个基因的快速策略也正在出现。
在某些情况下,由发现它们的实验室对某些基因进行分子诊断。
非综合征性视锥视杆细胞营养不良与其他色素沉着性视网膜病变的鉴别诊断
视锥视杆细胞营养不良通常与原发性外周视网膜病变和黄斑变性明显区分。 但是,CRD有时可能与几种临床实体共享特征。
视网膜色素变性
典型RP(杆体-锥体视网膜变性,RCD)。 在典型RCD中,诊断很容易,因为先进个症状是夜盲。 这个症状通常在正常视力的情况下独立存在几年,然后白天的视力损失变得突出。 在眼底中,色素沉积位于外周。
黄斑早期参与的RP。 在某些情况下,RCD有典型的缓慢进展,但黄斑参与相当早,出现一定程度的视力下降。 以夜盲为主和ERG上杆体受损为主的病史支持RCD的诊断。
早发型RP或晚期RP。 在与早发和严重RCD相关的病例中,黄斑参与的视力下降也可能发生得很早。 再次重要的是确定疾病过程中夜盲或中央视力丧失哪个体征出现得更早,并进行ERG。 当在晚期检查患者时,诊断可能特别困难。 那时,ERG中的典型改变无法检测。
Leber先天性青光眼(LCA)
该疾病与高度的视功能损害相关,这种损害在出生时就已经存在,表现为以杆体或锥体为主的疾病,或两者兼有。 眼肌阵挛、光固定和反应不良、视力低于1/20和平坦ERG是该疾病的主要体征。 与早发性CRD的鉴别诊断可能很困难,因为两种疾病有相同的临床体征。 几年后视功能残疾的剧烈恶化说明疾病应分类为CRD而不是LCA。
黄斑病变
扩展性大黄斑病变与晚期CRD或RP可能难以鉴别。在所有病例中,全视野ERG都是关键的检查。
斯塔格德氏病是一种外周视网膜通常没有病变的黄斑病变。该疾病很容易识别,表现为可能覆盖整个眼底的黄色斑点(眼底黄斑点)、荧光素血管造影中黄斑区超荧光病变(牛眼)和暗色素视网膜。然而,在一些晚期斯塔格德氏病例中有扩展性病变,此外,许多CRD是由“斯塔格德氏病基因”ABCA4引起的。在这些病例中,CRD的早期阶段可能类似于斯塔格德氏病,但在十年内会出现外周受累的体征。
锥体变性。在这些疾病中,杆体保持正常。主要临床体征是视力下降、光恐惧、失色觉和ERG上仅锥体受损。然而,在某些锥体变性中,杆体可能也有一定的受损,特别是在晚期。与视锥视杆细胞营养不良相反,在这些晚期阶段杆体至少部分得以保存,而在晚期CRD中则无法记录。另一个体征是多年没有黄斑病变,尽管视力下降。
视网膜静止性疾病
这本质上是无色觉,根据主要锥体受损(杆体并非全部正常)、缺乏疾病进展和正常眼底进行诊断。
遗传咨询
一旦确诊CRD,应告知患者并推荐进行家族调查。由于CRD中可能遇到所有遗传形式,遗传咨询总是被建议的。正确的表型诊断总是强制性的,在没有家族史或散发性病例中特别有用。
产前诊断
如果已经识别出负责的基因,则可以在家族中进行产前诊断。然而,产前诊断(羊膜穿刺或绒毛活检)引发了一个伦理问题:考虑到这种非危及生命的疾病,与这些侵入性产前程序相关的研究风险是否合理是值得质疑的。
看护和治疗
目前,还没有可以阻止色素沉着性视网膜病变进展或恢复视力的治疗。但是,有几种治疗策略旨在减缓退变过程(光保护、维生素治疗)、治疗诸如白内障、黄斑水肿、炎症等并发症,并帮助患者应对失明的社会和心理影响。事实上,视锥视杆细胞营养不良的管理与典型RP的管理没有区别。应特别关注过滤镜片以贼小化光恐惧和低视力辅助。患者常在20年内严重视力残疾或法定失明。因此,他们的教育应侧重于适应的职业(教学、基于计算机的活动、物理治疗师)很重要。
佳学基因将继续解决的问题
已克隆的基因只解释了一小部分自动主性视锥视杆细胞营养不良病例,可能一半的自动体隐性病例。因此,基因有待发现。理解编码蛋白的作用通常需要多年。如今,对于许多蛋白,有大量关于其功能的信息,而某些蛋白仍知之甚少。
一个具有挑战性的问题是阐明从基因突变到光感受器退行的确切步骤。动物模型和临床研究的数据表明,光感受器在整个生命周期内以线性速率发生凋亡(命名为“一次击中”假说),意味着它们有一定的发生凋亡的概率,这个概率从疾病早期到晚期保持不变。 对于某些基因或严重的突变,这个概率会很高,而对其他的会较低。实验和临床研究的结果清楚地表明,光感受器退行的机制是多重的。 在到目前为止研究的所有遗传性视锥视杆细胞营养不良中,数据不完整。 此外,凋亡途径可能同时涉及光感受器丢失,这也需要仔细研究。 这些知识对于设计治疗至关重要。 必须在动物模型和人体中证明各种潜在治疗的疗效。 例如,小鼠中RDS的基因替代治疗改善了光感受器的超微结构,但对光感受器细胞丢失没有显著影响。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
