












【佳学基因检测】3β-羟基类固醇脱氢酶缺陷症基因解码、基因检测
遗传病、罕见病基因检测导读:
根据《人体基因序列变化及其疾病表征》,3β-羟基类固醇脱氢酶缺陷症基因解码基因检测是采用超越数据库比对技术,对英文疾病名称为3-beta-hydroxysteroid dehydrogenase deficiency的疾病所进行的基于DNA序列变化而进行的临床医学基因检测。该病的其他英文名称包括:3 beta-HSD deficiency 3 beta-ol dehydrogenase deficiency、3-beta–hydroxysteroid dehydrogenase deficiency、3b-hydroxysteroid dehydrogenase deficiency、3β-HSD deficiency、3β-HSD deficiency congenital adrenal hyperplasia、3β-hydroxysteroid dehydrogenase deficiency、Type II 3β-hydroxysteroid dehydrogenase deficiency。 因此,3β-羟基类固醇脱氢酶缺陷症基因解码基因检测的项目名称有可能以以下几种形式出现:3β-HSD缺乏症基因筛查、3β-ol脱氢酶缺乏症基因测试、3-β-羟基类固醇脱氢酶缺乏症分子检测、3b-羟基类固醇脱氢酶缺乏症遗传性检查、3β-HSD缺乏症分子诊断检测、3β-HSD缺乏症先天性肾上腺增生症基因测序分析、3β-羟基类固醇脱氢酶缺乏症全外显子测序检查、II型3β-羟基类固醇脱氢酶缺乏症基因诊断。
3β-羟基类固醇脱氢酶 II 型缺乏症(HSD3B2 缺乏症)是一种罕见的先天性肾上腺增生症 (CAH),其特征是男性有不同程度的失盐和不有效男性化,女性有轻度男性化或外生殖器正常。 在佳学基因检测病案集中有一名染色体基因检测结果为46XY) 的病例,表现出盐分流失和男性化不有效、17OHP(17 羟孕酮)、ACTH(肾上腺皮质激素)、睾酮和 delta4 雄烯二酮 (delta4A) 水平显着升高、皮质醇水平低和骨质缺失和经常作为POR(细胞色素 P450 氧化还原酶)缺乏症的特征的骨骼改变。 通过 Sanger 测序对 HSD3B2 基因进行的突变分析表明,患者出现了两个新变体 c.370A>G p.Ser124Gly 和 c.308-6 G>A 的复合杂合子。 这两个 HSD3B2 基因变突变序列也存在于患者的哥哥身上,仅表现出不有效的男性化。 基因解码分析揭示了 c.370A>G p.Ser124Gly 的可能破坏作用:残基 p.Ser124 在物种间高度保守,似乎位于酶的催化位点,在 NAD(H) 中起关键作用 与其底物结合。 Intronic c.308-6G>A 变体预计可能致病; 替换似乎导致位于变体下游 6bp 的剪接受体位点发生变化。 这两个兄弟姐妹似乎受到 3β-HSD2 缺陷的影响; 然而,这两种新变体可能会导致该疾病在不同患者表现为不同的疾病表征。
什么样的人应该做3β-羟基类固醇脱氢酶缺陷症基因解码、基因检测?
3-β (β)-羟基类固醇脱氢酶 (HSD) 缺乏症是一种遗传性疾病,会影响产生激素的腺体,包括性腺(女性的卵巢和男性的睾丸)和肾上腺。 性腺在出生前和青春期指导性发育。 位于肾脏顶部的肾上腺调节某些激素的产生并控制体内的盐含量。 缺乏 3β-HSD 的人缺乏这些腺体中产生的许多激素。 3β-HSD 缺乏症是一组称为先天性肾上腺增生的疾病之一,会损害激素的产生并破坏性发育和成熟。
3β-HSD 缺乏症分为三种类型:失盐型、非失盐型和非典型型。 在失盐型中,激素的产生极低。 患有这种类型的人会在尿液中流失大量的钠,这可能会危及生命。 由于缺乏盐重吸收相关的并发症,包括脱水、进食不良和呕吐,患有失盐型的个体通常在出生后不久就被诊断出来。 患有非耗盐型 3β-HSD 缺乏症的人会产生足够的激素,以允许肾脏重吸收钠。 非经典型患者的症状最轻,不会出现盐消耗。
在患有任何类型 3β-HSD 缺乏症的男性中,雄性激素问题会导致外生殖器异常。 这些异常的范围从尿道开口位于阴茎下侧(尿道下裂)到外生殖器看起来不是男性或女性(不明确的生殖器)。 生殖器异常的严重程度并不总是取决于疾病的类型。 由于睾丸激素功能障碍,患有 3β-HSD 缺陷的男性通常无法生育(不育)。
患有 3β-HSD 缺乏症的女性在出生时可能会出现外生殖器轻微异常。 受非耗盐型或非经典型影响的女性通常要到童年中期或青春期才被诊断出来,那时她们可能会出现月经不调、阴毛过早生长和体毛过度生长(多毛症)。 患有 3β-HSD 缺乏症的女性很难怀孕(生育能力受损)。
3β-羟基类固醇脱氢酶缺陷症的致病基因鉴定基因解码:基因检测的科学依据
HSD3B2基因的变异导致3β-HSD缺陷,该基因编码制造3β-HSD酶的指令,该酶存在于生殖腺和肾上腺中。3β-HSD酶参与合成多种激素,包括皮质醇、醛固酮、雄激素和雌激素。皮质醇有多种功能,如维持能量和血糖水平、保护身体免受压力和抑制炎症。醛固酮有时被称为“保钠激素”,因为它调节肾脏保留的盐量,盐的保留影响液体水平和血压。雄激素和雌激素对于正常的性发育和生殖是必不可少的。
3β-HSD缺陷是由于缺乏3β-HSD酶而导致的。功能性酶的数量决定一个人是否患有盐浪费型或非盐浪费型障碍。患有盐浪费型的人具有HSD3B2基因突变,导致酶的产生非常少或没有。而患有非盐浪费型的人则具有HSD3B2基因突变,其允许产生一些功能性酶,但数量减少。
3β-HSD是肾上腺和生殖腺中合成类固醇的关键酶之一。该酶是一种42-kDa的微粒体酶,能将羟基转化为碳3上的酮基,并将Δ5类固醇前体异构化为Δ4酮类固醇。
3β-HSD将孕烯醇酮转化为黄体酮,将17β-羟基孕烯醇酮转化为17β-羟基孕酮,将脱氢表雄酮(DHEA)转化为雄烯二酮,将雄烯二醇转化为睾酮。HSD3B2和HSD3B1是位于1号染色体(1p13.1)上的两个密切相关的基因,分别编码II型和I型同工酶。HSD3B1主要表达于胎盘和外周组织,而HSD3B2主要表达于肾上腺和生殖腺。
3β-HSD2缺乏症 (OMIM # +201810)是一种罕见的先天性肾上腺增生疾病,由HSD3B2基因的有害突变引起,呈常染色体隐性遗传。患病率为1:1,000,000,与肾上腺和性腺中类固醇合成的损害有关。 典型的3β-HSD2缺乏症会导致肾上腺合成糖皮质激素和盐皮质激素的功能障碍,以及过量的Δ5-类固醇,导致肾上腺功能不全,可能伴随失盐症状。
除肾上腺功能障碍外,3β-HSD2缺乏症患者的性腺功能也会受到影响。受影响的男性个体可能出现不有效男性化,临床症状可能从轻度的尿道下裂到由于睾酮生物合成受损导致的生殖器不明确不等。46XX个体可能表现出正常的女性生殖器或中度的外生殖器男性化迹象,并伴有阴蒂肥大。这些特征与胎儿肾上腺过度产生DHEA有关,DHEA可以通过肾上腺外的3βHSD1转化为睾酮。
因此,外周3β-HSD1活性的存在常常使该疾病的激素诊断变得复杂。
3β-羟基类固醇脱氢酶缺陷症基因检测大数据分析
根据《佳学基因检测大数据分析》, 3β-羟基类固醇脱氢酶缺陷症的患病率为百万分之一。
佳学基因是如何做3β-羟基类固醇脱氢酶缺陷症的致病基因鉴定的?
佳学基因的3β-羟基类固醇脱氢酶缺陷症的致病基因鉴定可能是通过以下步骤完成的:
1.临床表型特征鉴定:医生可以首先对患者进行身体检查和症状评估,包括肾上腺功能不全和生殖器发育不良等特征,以确定患者是否患有3β-羟基类固醇脱氢酶缺陷症。
2.遗传学分析:对患者进行基因检测,检测是否存在3β-羟基类固醇脱氢酶缺陷症相关的致病基因突变。佳学基因采用的基因解码技术通常是进行全外显子测序,以避免因疾病表征未有效展示而出现的误诊,并尽量实现更为早期、更为明确的诊断。高通量测序后的诊断结果,佳学基因还采用一代测序的方法对检测结果进行多次验证。通过突变在父母中的存在与否,结合遗传规律对检测结果进行多重验证。
3.功能验证:如果在患者中发现了与3β-羟基类固醇脱氢酶缺陷症相关的致病基因突变,而这些突变缺乏数据库、病例记录支撑。佳学基因一般会委托基础科研机构进一步进行功能验证来确认这些突变是否会导致疾病的发生。这可以包括利用细胞模型或动物模型进行相关的分子和细胞水平的实验。这一额外步骤使得佳学基因的合作机构可以在该类疾病的诊断和治疗方面处于国际领先地位。
综合以上步骤,佳学基因的3β-羟基类固醇脱氢酶缺陷症的致病基因鉴定基因检测可以确定疾病发生的原因,并为该疾病的治疗、预防和出生缺陷规避提供指导。
3β-羟基类固醇脱氢酶缺陷症基因检测案例
3β-羟基类固醇脱氢酶缺陷症基因检测的临床观察及常规检验
患者n.1
患者 n.1 是一名足月新生儿(胎龄 = 41 周)在自然阴道分娩时平安无事地出生。 出生体重和身长分别为3330 g和51.5 cm。 Apgar 评分在 1 分钟和 5 分钟时分别为 9 分和 9 分。 该患者表现出色素沉着过度和会阴部尿道下裂:在严重色素沉着过度的双裂阴囊中可触及 2 mL 的两个睾丸。 在 3 天大时,受检患儿接受了荷尔蒙异常筛查:测试显示 17OHP 水平显着升高(86 ng/mL n.v. 10 ng/mL),ACTH 250 pg/mL(n.v. 8–53),皮质醇 42 µg/L (n.v. 60–230),和 DHEAS 1970 mcg/L (560–2360)。 睾酮和 delta4 雄烯二酮 (delta4A) 分别为 1.3 ng/mL(n.v. <0.4)和 18.5 ng/mL(n.v. 0.60–5.60)。 肾素、醛固酮和电解质在正常范围内。
染色体基因检测提示核型为46 XY。 鉴于先天性肾上腺增生症中存在肾上腺功能不全,开始使用初始剂量为 35 mg/mg/天的氢化可的松进行替代治疗。 在 18 天大时,患者表现出一些盐消耗迹象并伴有轻度低钠血症 (Na = 130 mEq/L); 因此,在治疗中加入氟氢可的松 0.05 mg/天和口服 NaCl 补充剂 1 g/天。
临床医师决定对患者进行 HSD3B2 基因突变检测,以调查在没有骨骼改变的情况下出现盐分丢失和不有效男性化的可能遗传原因,这些骨骼改变在 POR 缺陷中更常见。 在出生后第三天测得的高水平睾酮 (1.3 ng/mL) 应该反映出母体荷尔蒙的干扰。
患者 n.2
患者 n.2 是患者 n.1 的哥哥。 在进行临床和生化筛查时,已经是 2.5 岁。 他是一名足月新生儿(胎龄 = 40 周),在顺利通过阴道分娩后出生。 出生体重为3110克,身长为51.5厘米。 Apgar 评分在 1 分钟和 5 分钟时分别为 10 分和 10 分。 神经系统检查正常。 没有报告乏力或其他健康问题。 体重为 12.5 公斤(-0.56 SDS),身高为 89 厘米(-039 SDS)。 现出与兄弟相同的严重会阴尿道下裂,有进行手术矫正的需要,双侧阴囊性腺正常 2.5 mL(青春期 G1PH1)。 新生儿色素沉着过度由父母转诊。 激素数据未显示肾上腺功能不全
3β-羟基类固醇脱氢酶缺陷症基因检测结果
HSD3B2 基因的测序显示患者 n.1 和他的兄弟(患者 n.2)携带两个以前从未描述过的突变:c.370A>G p.Ser124Gly 和 c.308-6 G>A(图 1a –d)。父母双方都是临床健康的突变杂合子携带者; 父亲携带错义突变,而母亲携带内含子突变(图 1e-f)。
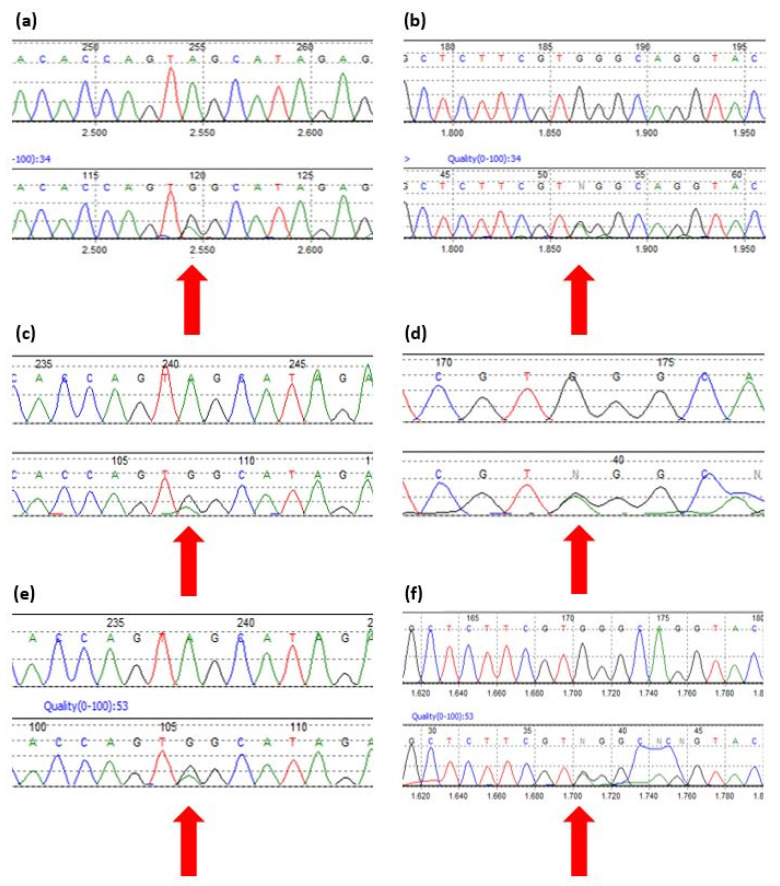
图1:来自 Mutation Surveyor v3.30 的序列报告了在患者 n.1、患者 n.2 及其父母中发现的突变。 (a) 患者 n.1 错义突变 HSD3B2 c.370 A>G。 (b) 患者 n.1 内含子突变 HSD3B2 c.308-6 G>A。 (c) 患者 n.2 错义突变 HSD3B2 c.370 A>G。 (d) 患者 n.2 内含子突变 HSD3B2 c.308-6 G>A。 (e) 父亲的错义突变 HSD3B2 c.370 A>G。 (f) 母亲的内含子突变 HSD3B2 c.308-6 G>A。
3β-羟基类固醇脱氢酶缺陷症基因检测论
佳学基因病案集分享了儿童医院儿科内分泌学中心就诊的两个同胞兄妹的病史,他们在 HSD3B2 基因上出现了两个杂合突变,这两个突变未被以前的数据库收录。 患者 n.1 表现出更严重的情况,显示出肾上腺激素谱的改变以及色素沉着过度和会阴尿道下裂。 另一方面,虽然 2 号患者的激素测试未显示肾上腺功能不全,但他们仍然表现出严重的会阴尿道下裂。
在两个兄弟姐妹中发现的第一个变异体遗传自健康的父亲,位于外显子 4 (c.370 A>G),导致错义突变,导致第 124 位丝氨酸变为甘氨酸。 致病基因鉴定基因解码揭示了这种基因突变可能的破坏作用:残基 p.Ser124 在物种间确实高度保守,似乎位于酶的催化位点,在 NAD(H) 与其底物结合中发挥关键作用 [ 20]。 实际上,类固醇前体转化过程中的第一个反应是脱氢酶将 3β-羟基氧化成酮,在此过程中 NAD+ 被还原为 NADH。 中间体 Δ5,3-酮类固醇仍然与具有新生 NADH 的酶结合,辅因子结合位点中实际存在的 NADH 导致 Δ5-Δ4-异构酶活性的激活。
第二个变体在内含子 3 (c.308-6 G>A) 上发现,遗传自健康的母亲。 根据 ACMG 的建议,由于在 gnomAD 人口数据库中频率极低,内含子变体被归类为具有以下等级的 VUS:PM2(中等)。 尽管如此,基因解码分析揭示了一种可能的致病效应,因为将鸟嘌呤替换为腺嘌呤似乎形成了一个新的二核苷酸,导致位于变体下游 6bp 的剪接受体位点发生变化,从而导致移码或激活 一个新的隐蔽位点,可能导致一种无功能的酶。
由于这种疾病的罕见性,表型-基因型相关性尚未在大型病例系列中得到充分研究。 尽管一般来说,功能和生化数据与表达的表型一致,但受影响患者的临床表型明显异质; 此外,在表现出不同临床结果的患者的 HSD3B2 基因中发现了相同的 DNA 变体 [3,21,22]。 其中,在 Ladjouze 等人最近的一项工作中,作者报告了相同突变 (c.665C>A) 的发现,这两种突变均发生在表现出失盐形式的患者和表现出非失盐形式的患者中。
- 【佳学基因检测】拉伦综合症基因检测的坑不要跳!...
- 【佳学基因检测】如何确定格兰奇综合症(GRNG)基因检测的质量?...
- 【佳学基因检测】肉芽肿性多血管炎(GPA)基因解码分析法...
- 【佳学基因检测】灰血小板综合征一定要做基因检测吗?...
- 【佳学基因检测】怎么检查高登哈综合征基因?...
- 【佳学基因检测】戈林·乔杜里·莫斯综合症基因测序基因检测...
- 【佳学基因检测】GLA缺乏症可以做基因检测吗?...
- 【佳学基因检测】吉尔·德拉图雷特综合征基因检测如何理解?...
- 【佳学基因检测】吉莱斯皮综合征要做基因检测吗?...
- 【佳学基因检测】得了生长激素受体缺乏症还能生孩子吗?...
- 【佳学基因检测】家族性水平性凝视麻痹,伴有进行性脊柱侧弯一定要做基因检测吗?...
- 【佳学基因检测】GAMT 缺陷基因检测的可信度?...
- 【佳学基因检测】戈谢综合症基因检测数据库比对与结构功能分析法...
- 【佳学基因检测】先天性眼外肌纤维化的临床诊断标准...
- 【佳学基因检测】系统性硬皮病基因检测如何做?...
- 【佳学基因检测】怎么知道是否遗传了多发性内分泌肿瘤基因?...
- 【佳学基因检测】2 型神经纤维瘤病基因测序基因检测...
- 【佳学基因检测】福克斯内皮营养不良要做基因检测的基本知识...
- 【佳学基因检测】想了解弗雷泽综合症基因及弗雷泽综合症基因检测...
- 【佳学基因检测】想了解法伯氏病基因及法伯氏病基因检测...
- 【佳学基因检测】家族性匹克氏病为什么要做基因检测?...
- 【佳学基因检测】法布里氏病基因检测标准如何确定的...
- 【佳学基因检测】在医院做二氢嘧啶脱氢酶缺乏症基因检测是怎样的?...
- 【佳学基因检测】肺泡毛细血管发育不良伴肺静脉错位怎么做基因检测?...
- 【佳学基因检测】新生儿筛查就是新生儿基因检测吗?...
- 【佳学基因检测】新生儿筛查出苯丙酮尿症为什么还要做基因检测?...
- 【佳学基因检测】新生儿筛查是什么项目?可以替代基因检测吗?...
- 【佳学基因检测】儿科常见的遗传病有哪些?怎么做基因检测?...
- 【佳学基因检测】全基因组测序(WGS)基因检测如何变靶罕见病的诊断...
- 【佳学基因检测】枫糖尿病是一种单基因遗传病...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
