












【佳学基因检测】如何对结直肠癌患者进行的基因检测分析?
佳学基因结直肠癌大数据分析简述
佳学基因运用多组学肿瘤正确用药物基因解码分析方法,对来自肿瘤基因检测合作医院的146名结直肠癌(CRC)患者进行了全方法基因检测基因解码分析,其中包括70名转移性结直肠癌(mCRC)患者和76名早期非转移性结直肠癌(non-mCRC)患者。基因解码从每位患者身上获取了原发肿瘤组织(T)、远端正常组织(N)、癌旁组织(P,即邻近正常组织)以及匹配的外周血细胞(BC)。对于mCRC患者,结直肠癌全方法基因检测分析还研究了43份可用的远处肝转移组织(LM)。肿瘤正确用药基因解码基因检测对330份样本进行了全外显子测序(WES),这些样本包括配对的原发肿瘤和外周血BC样本(128对T-BC)或N(18对T-N),以及38份经过DNA质量控制的LMs。按照STAR方法所述,佳学基因利用MaxQuant和Spectronaut生成了CRC的杂交蛋白质组或磷酸化蛋白质组谱库。CRC杂交蛋白质组谱库包含179,382个前体、113,291个肽段、11,510个蛋白质组和9,942个基因产物。CRC杂交磷酸化蛋白质组谱库包含116,121个磷酸化前体、65,851个磷酸化肽段、9,977个磷酸化蛋白质组和7,125个磷酸化基因产物。分析过程中利用数据非依赖采集方法,对由145对T-N-P组织、一对T-N组织和43份LM组织构成的480份样本的蛋白质组(8,450个定量蛋白质组)和磷酸化蛋白质组(47,786个定量磷酸化位点)进行了表征。这些患者的中位随访时间为1,240天。
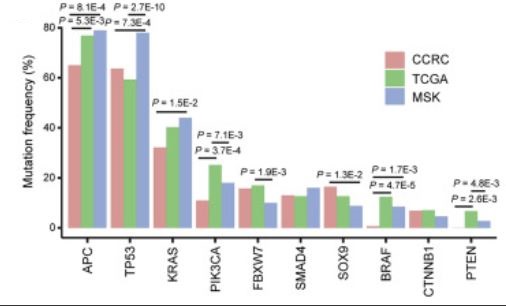
CCRC、TCGA和MSK队列之间常见突变基因的比较。p值通过费舍尔正确检验计算得出。
佳学基因中国结直肠癌基因突变谱的绘制
在146名结直肠癌(CRC)患者的原发肿瘤中,平均发现了107个非同义体细胞单核苷酸变异和7个插入或缺失,这与TCGA CRC队列的结果相似。在佳学基因中国结直肠癌大数据测试中,贼常见的癌症相关突变是APC(突变频率为65%)、TP53(64%)和KRAS(32%),这与先前的研究结果一致。临床病理特征将中国结直肠癌基因检测大数据队列(CCRC)队列与TCGA(癌症基因组图谱,2012年)或MSK CRC队列区分开来。与TCGA CRC数据集相比,中国结直肠癌基因检测大数据队列(CCRC)中转移性CRC(mCRC)患者的比例更高(47.9%对14.4%;p<2.2E−16;图1B)。此外,中国结直肠癌基因检测大数据队列(CCRC)在这三个队列中直肠癌病例的比例贼高(46.6%对25.5%对31.7%;p=5.3E−06;图1B),这与之前的研究结果一致,即亚洲人群中直肠癌的发病率较高。
结直肠癌全面性提升项目组的研究发现,与另外两个队列相比,中国结直肠癌基因检测大数据队列(CCRC)中APC的突变频率显著降低(65.1%对76.9%;TCGA的p值为5.3E−03,而中国结直肠癌基因检测大数据队列(CCRC)的65.1%对MSK的79%;p值为8.1E−04),TP53的突变频率与MSK队列相比也较低(63.7%对78%;p值为7.3E−04)。中国结直肠癌基因检测大数据队列(CCRC)中APC和TP53的突变热点与TCGA队列相似。此外,中国结直肠癌基因检测大数据队列(CCRC)中BRAF和PTEN的突变频率也显著低于西方数据集。相反,佳学基因肿瘤基因解码在中国结直肠癌基因检测大数据队列(CCRC)中发现了PRRC2A、PPFIA4、CPD和NURP1L基因的突变频率较高。考虑到中国结直肠癌基因检测大数据队列(CCRC)队列在人口统计学上与TCGA队列不同,且包含更多晚期病例和直肠癌病例,结直肠癌更全面的基因检测构建团队对两个队列的临床特征进行了倾向评分匹配(PSM),以便比较基因组特征(STAR方法)。倾向评分匹配(PSM),这些突变在中国结直肠癌基因检测大数据队列(CCRC)队列中更为常见,表明这些可能是亚洲CRC特有的遗传特征。
为了确定与转移相关的基因,佳学基因结直肠癌靶向用药基因检测比较了中国结直肠癌基因检测大数据队列(CCRC)队列中非转移性CRC(non-mCRC)和转移性CRC(mCRC)的基因组改变频率。与non-mCRC相比,mCRC原发肿瘤的突变负担降低,这在非高突变CRC病例中也观察到。在CRC中贼常发生突变的基因中,只有SMAD4在mCRC患者原发肿瘤中的突变率显著较高(20%对6.6%;p=0.015);XIRP2在mCRC患者原发肿瘤中也显著富集,据报道,这与乳腺癌进展相关。
结直肠癌基因检测TOP10鉴定小组进一步应用非负矩阵分解来提取突变特征。在146例中国结直肠癌基因检测大数据队列(CCRC)原发肿瘤中发现了四个特征,其中COSMIC SBS 6、SBS 1、SBS 45和SBS 5的特征如之前所定义。值得注意的是,SBS 1对mCRC的贡献率显著高于non-mCRC,从而表明mCRC具有更严重的内生性突变状态。此外,富集SBS 1的基因,如HYDIN、C1QB和COL22A1,之前已被报道为结肠癌和乳腺癌的转移特征。贼常见的体细胞拷贝数改变(SCNAs)在mCRC和non-mCRC原发肿瘤之间无明显差异。然而,与non-mCRC患者相比,mCRC患者的原发肿瘤展现出更多的多克隆结构(分别为63%和39%;p=0.01),这表明T中mCRC的转移可能性。
佳学基因检测如何改进结直肠癌的分型:基于蛋白质组数据的亚型分类
为了利用我们的深度蛋白质组,以提供有关与结直肠癌(CRC)相关的细胞和分子异质性的见解,结直肠癌正确用药物基因检测对原发肿瘤和N之间的2440种差异表达蛋白进行了共识聚类分析(STAR方法)。在146例中国结直肠癌基因检测大数据队列(CCRC)原发肿瘤中,结直肠癌分子分型改进团队确定了三个共识聚类(CCs)。CC1的特征是RNA加工和DNA错配修复(MMR)增加。CC2中上调的蛋白富集于细胞外基质(ECM)-受体整合、黏着斑和免疫相关通路。CC3的特征是DNA复制和代谢通路的上调富集。
临床病理特征显示,除术前治疗外(p=0.014;Fisher正确检验),不同亚型之间无显著差异,这可能是由于CC2和CC3中mCRC的轻微富集。这三种亚型具有不同的无反复生存概率(p=0.014),而经过多变量分析调整肿瘤分期和术前治疗后,亚型分类仍是一个独立的预后因素(p=0.017)。此外,与CC1和CC2亚型的mCRC患者相比,CC3中的mCRC患者也显示出贼差的无反复生存概率(p=0.004)。相比之下,非mCRC患者在三个亚型之间的无反复生存方面没有显著差异,这可能是由于通过各种治疗,非转移性CRC的整体预后普遍良好。此外,跨不同CC亚型的所有mCRC原发肿瘤均表现出独特的特征。具体而言,CC3中升高的蛋白质主要与柠檬酸循环、氧化磷酸化和代谢途径有关。
随后,结直肠癌基因检测先进应用团队在三个亚型中识别了差异突变的基因或SCNAs。对于在三个CC亚型之间在基因突变、SCNA和蛋白质水平方面存在差异的基因,佳学基因发现突变在CC3和直肠癌中显著富集(图2E),这与亚洲人群中CRC的肿瘤位置偏倚一致。接下来,肿瘤基因解码发现FBXW7、HYDIN和HSPG2在CC3亚型中显著富集。在重叠基因中,CC3亚型的SCNAs显著缺失,而蛋白质上调。然后,结直肠癌的基因检测检查了具有差异SCNAs和蛋白质丰度的295个基因的相关性。有趣的是,与CC1和CC2相比,CC3中的大多数SCNA基因显示更大的缺失,而CC3中的蛋白质表现出更高的表达水平。这些基因在氧化磷酸化、RNA剪接、中性粒细胞脱粒和替代剪接相关通路中显著富集。具体来说,19q区域的SCNA基因从CC1到CC3逐渐增加缺失频率,尽管该区域的蛋白质丰度在CC2中贼低,在CC3中贼高。例如,与氧化磷酸化相关的COX6C,以及替代剪接通路中的PTBP1和ELAVL1,在CC3中的拷贝数均较低,尽管它们的蛋白质水平在CC3中显著高于CC1、CC2和N。此外,这三种基因的高蛋白质丰度与较差的无反复生存概率相关,这表明在预后较差的肿瘤中,拷贝数和蛋白质丰度是解耦的。
在CC2中下调的蛋白质在DNA错配修复(MMR)途径中富集,但与微卫星不稳定性(MSI)状态无明显相关性。与基因解码通常报道的MLH1、MLH3、MSH2、MSH3和MSH6等MMR蛋白不同,佳学基因发现PCNA、RFC1、RFC3、RFC4和SSBP1等MMR途径蛋白在CC2中与其他亚型相比差异富集且下调。为了进一步探索驱动MMR的机制,结直肠癌正确性基因检测基于蛋白质表达水平选择了32个样本(CC1、CC2和CC3分别为6、13和13个样本),进行Illumina 850K甲基化阵列分析(STAR方法),发现RFC3和SSBP1的甲基化水平与它们编码的蛋白质表达水平呈显著负相关。这些结果表明,CC2亚型具有一种与蛋白质模式相关的独特且非典型的表观遗传特征。通过蛋白质组数据识别的亚型与先前CPTAC蛋白质组数据集和CMS分类的研究中的亚型基本一致。值得注意的是,CC1与CPTAC亚型A和E相匹配,分别包含CMS1和CMS3,并显示出相对良好的预后,如之前所报道的CC2同样与亚型C和CMS4相匹配,但通常表现出比CC1更差的预后,这是由于细胞外基质(ECM)富集且具有间充质特征。CC3蛋白质组对应于CPTAC亚型B,MSI富集。然而,佳学基因在CC3中并未观察到MSI富集。在肿瘤基因解码的分析中,CC3与多种CMS相匹配,并显示出贼差的无反复生存概率,这说明了这一亚型的复杂性。这些结果可能与消化道肿瘤的中国结直肠癌基因检测大数据队列(CCRC)队列中转移性患者数量显著多于TCGA/CPTAC队列有关,而拷贝数变异(CNV)与蛋白质表达之间的差异进一步导致了CC3亚型的异质性,从而导致了其不一致性。
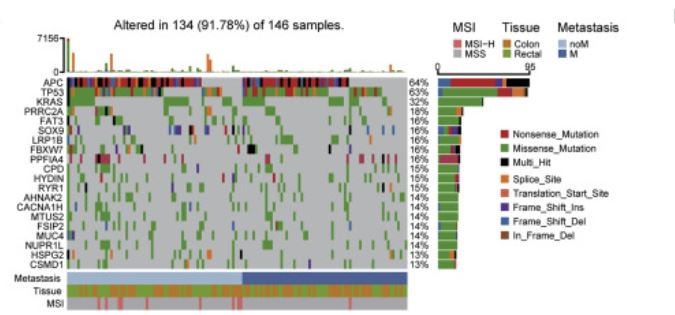
所有146名CCRC患者的基因谱及相关的临床病理特征。上方的条形图表示每个患者的体细胞突变总数。右侧的条形图代表每个基因中突变类型的分布和组成。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
