












【佳学基因检测】基因解码基因检测的基于信号网络的基因数据分析法
基于网络分析的基因解码介绍:
由于癌基因的正确鉴定具有巨大的意义,并有助于揭示肿瘤的生物学行为,因此人们致力于减轻检测到的癌基因的不确定性。在本文中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队提出了一个基于差异网络的框架来检测具有生物学意义的癌症相关基因。首先,提出了一种基因调控网络构建算法,该算法采用基于似然得分和信息先验的boosting回归来提高识别的正确性。其次,利用该算法,从病例样本和对照样本独立构建了两个基因调控网络。第三,通过将两个网络相减,得到一个差异网络模型,然后用于对差异表达的枢纽基因进行排序,以识别癌症生物标志物。t -test 和 lasso),该方法在合成数据集和两个真实乳腺癌数据集上的正确性都有显着提高。此外,通过文献挖掘、GO分析和通路功能富集分析,对确定的6个乳腺癌易感基因( TSPYL5、CD55、CCNE2、DCK、BBC3和MUC1 )进行了验证。在这些癌基因中,TSPYL5和CCNE2已被称为乳腺癌的预后生物标志物,CD55已从文献证据怀疑在乳腺癌预后中起重要作用,而其他三个基因是新发现的乳腺癌生物标志物。更一般地说,差异网络模式可以扩展到其他复杂疾病,以检测疾病相关基因。
1.基因检测的网络分析基因解码
治疗癌症相当困难,因为越来越多的证据表明,癌症是一种复杂的遗传疾病,涉及多个基因、蛋白质、通路和调控相互联系。为了为癌症治疗提供有用的信息,进行了几项具有里程碑意义的研究 ,以揭示癌症发展、进展或反复的癌基因或生物标志物。
近年来出现了基于基因的方法来识别肿瘤相关基因集,例如 中定义的“自上而下”方法或 76 基因鹿特丹特征中的“贼小生物输入”。这些方法通常利用微阵列基因表达谱技术和差异表达分析来识别癌症相关基因,这些基因的表达水平在癌症患者中发生显着变化。尽管它们已被应用于识别与癌症发展或进展相关的生物标志物,但基于基因的方法经常受到巨大候选基因的不确定性的影响,这限制了鉴定癌症发生的致病基因和风险基因的信号网络分析团队对肿瘤出现和生长方式的理解。
为了识别复杂的相互作用模式、途径和过度代表的生物过程,基因集富集分析 (GSEA) 在基于基因的方法中被反复利用。GSEA 侧重于共享共同生物学功能或信号通路的基因组,这些基因分别由基因本体论 (GO) 或 KEGG 等定义。贼近的工作还表明,基于 GO 分析和通路信息检测到的生物标志物比单个标志物基因更具可重复性 。与原来的 70 个基因相比,这些生物标志物还可以将分类正确率提高 8% 。
越来越多的证据表明,癌症相关基因通常被组织成通路或基因网络,这些通路或基因网络由一组分子水平的相互作用基因组成。此外,从先前研究中发现的基因特征通常富含常见的癌症相关途径和类似的生物过程。许多研究人员似乎提倡和接受这样一种观点,即只有那些能够显着丰富肿瘤诱导的信号通路或相关生物学过程的分子诊断才对分子诊断有用和有价值。
已经提出了几种基于网络的方法来识别新的癌基因、子网络或参与肿瘤进展的途径。庄等人。 应用基于蛋白质网络的方法通过从蛋白质相互作用数据库中提取子网络来识别生物标志物。他们还证明,使用基于网络的方法检测到的生物标志物比没有网络信息选择的单个标志物基因更具可重复性。吴等人。 整合不同类型的网络和已知的基因-表型关联信息来计算相似性评分和预测疾病基因。弗罗利希 通过在蛋白质相互作用网络上映射不同的基因签名构建共识签名,其中基于蛋白质-蛋白质相互作用网络中不同基因的贼短路径距离执行聚类算法。此外,陈等人。 开发了一种用于癌症生物标志物识别的网络约束支持向量机方法。该方法通过整合基因表达数据和蛋白质-蛋白质相互作用数据,提高了网络生物标志物的预测性能。
差分网络分析在阐明基本生物反应以及发现不同生物状态之间的重要差异方面起着关键作用 。与传统的基于基因的方法相比,通过执行差异网络分析,可以识别出更多已知与疾病发展相关的特征基因或子网络。Valcarcel 等人。从空腹血糖正常(NFG)和空腹血糖受损(IFG)的男性中推断出一个差异网络,其中对网络进行了偏相关的收缩估计,然后利用统计检验探索了两者之间的差异。两个定义的组(NFG 和 IFG)。甘巴德拉等人。 开发了一个名为 DINA 的强大程序,使用稍微修改的信息熵测量来识别组织特异性途径。尽管它可以发现一组网络之间的差异,但 DINA 无法检测具有相同密度的不同网络拓扑。扬库等人。 揭示了基因共表达模式并使用自定义差分网络分析程序检测模块,包括相关系数、聚类和排列测试。此外,韦斯特等人。提出了差分网络熵,并证明正常组织和癌组织之间的基因表达差异与局部网络熵的变化是反相关的。这些发现可能对识别新的癌基因具有潜在意义。
在本文中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队提出了一种新颖的基于差分网络的推理框架,称为基于网络的统计分析方法 (netSAM) 来检测癌基因。使用差异网络建模和功能富集分析,而不是单纯的单个基因或通路的差异表达分析,netSAM 克服了基于基因的方法的一些局限性,例如识别的不确定性或不适合泛化。通过大量实验在模拟数据和真实数据上证明了netSAM算法的适用性和有效性。鉴定癌症发生的致病基因和风险基因的信号网络分析团队的结果表明,netSAM 优于两种基于基因的方法(t-test 和 lasso)在正确度、精度和重叠率等方面。此外,鉴定癌症发生的致病基因和风险基因的信号网络分析团队应用 netSAM 从两个基准数据集(Wang 等人和 Van De Vijver 等人)中识别乳腺癌基因,并获得了由 6 个基因(TSPYL5、CD55、CCNE2、DCK、BBC3、和MUC1),已通过 GO 和通路分析证明其在生物学上是合理的。文献挖掘表明,与以前的工作相比,得到的特征具有更高的预测能力,在预测乳腺癌转移和促进治疗决策方面都有用。
鉴定癌症发生的致病基因和风险基因的信号网络分析团队在本文中的贡献包括三个方面。首先,提出了一种新的基因调控网络构建算法,并正确高效地展示了其推理能力。第二个重要贡献是基于属性的无标度信息先验分数。第三,该方法的另一个重要贡献是用于识别癌基因的差异网络模式。这个框架可以很容易地扩展到其他复杂的疾病。
在本文的其余部分安排如下。在第 2 节中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队提供了 netSAM 的所有详细信息。第 3 节介绍了实验结果和分析。结论和未来的工作可以在第 4 节中找到。
2. 材料和方法
2.1 基于差分网络的推理框架
鉴定癌症发生的致病基因和风险基因的信号网络分析团队提出了一种新的基于差分网络的方案netSAM,以基于整个网络的连锁特征来评估基因的相对重要性。首先,netSAM 通过过滤差异表达的基因以及从“病例”和“对照”样本中推断差异网络来探索不同癌症表型的转录调控机制。其次,netSAM 选择得分贼高的相互作用基因,这些基因似乎构建了癌症相关的子网络,作为癌症易感性的候选基因。在这个过程中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队假设一个基因的得分越高,它就越有可能是癌症相关基因。贼后,鉴定癌症发生的致病基因和风险基因的信号网络分析团队研究了排名靠前的基因的功能富集并评估了生物标志物的高效性。
与基于基因的方法相比,netSAM 的优点或特点包括(a)通过构建差异网络而不是差异表达分析来识别癌基因,(b)专注于提供对功能模块的洞察的“中心”基因或途径,和(c)通过网络推断以及无标度网络的特征揭示基因调控关系。
一般来说,癌症基因的基于差异网络的检测包括五个步骤,如图1.
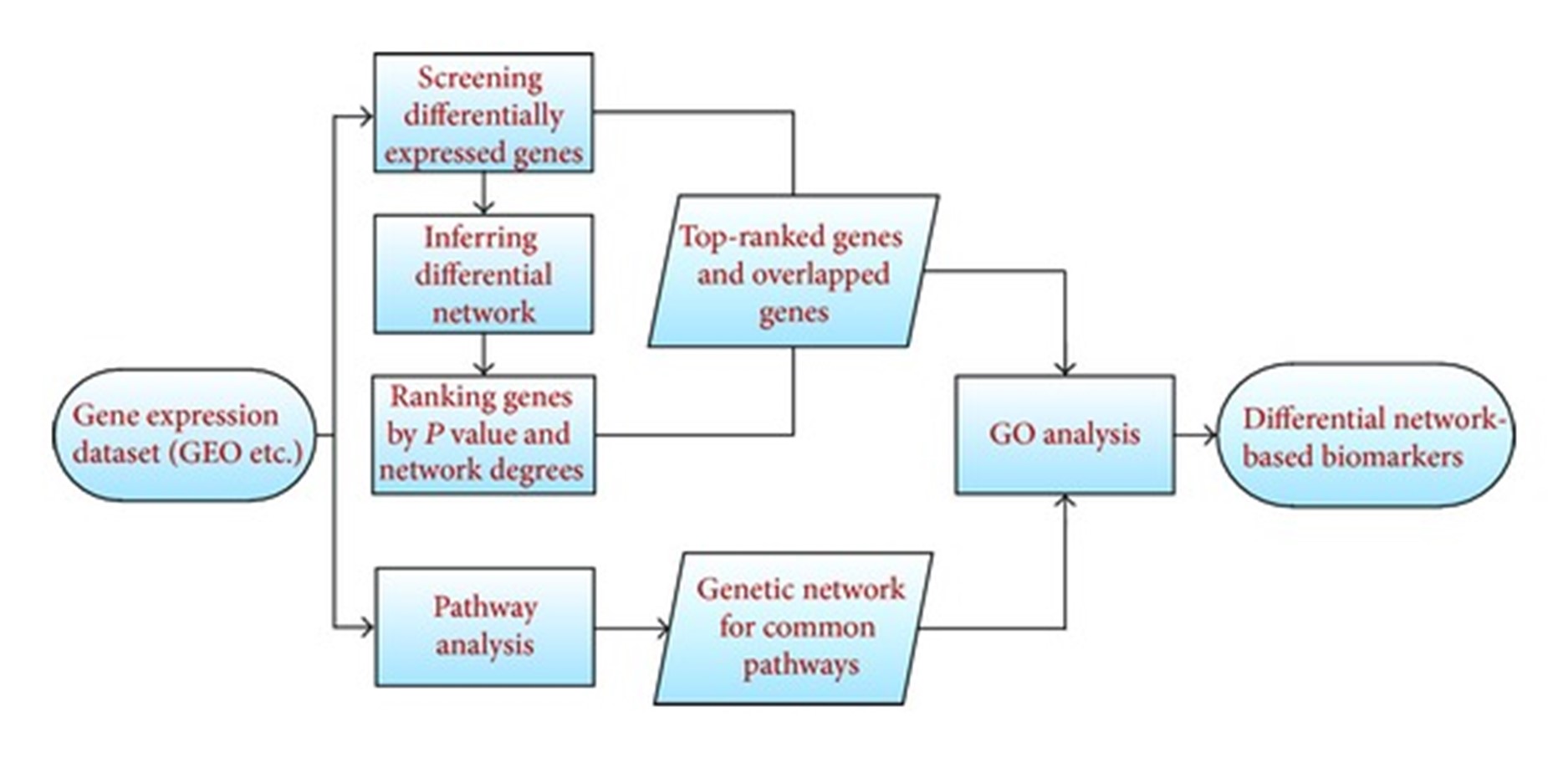
图1:方案流程图:基于差异网络的癌症生物标志物识别。
2.2. 贝叶斯准则和后验分数
所有系数。
2.3. 候选基因的功能富集分析
基因集富集分析 (GSEA) 是一种计算工具,用于研究预定义的基因集是否显示出统计学意义。构建包含基因本体的生物学过程术语的基因集,然后通过使用 Cytoscape 的 BiNGO 插件进行 GO 分析,在检测到的癌症基因特征中研究过度表示的 GO 类别。采用基因本体功能富集分析,其中超几何检验用于功能过度表示和错误发现率用于多假设检验校正。只有小于 0.05的校正P值被认为是显着的。
此外,研究了差异遗传相互作用与已知途径之间的关联。如差异网络所示,差异遗传相互作用更可能发生在连接两个不同子网络的基因对之间,而不是同一子网络内的基因对之间。在这些发现的基础上,构建了基因及其差异遗传相互作用的图谱,其中一些中心以前没有与癌症发展相关联。为了验证新发现的癌基因,使用 DAVID 进行通路分析,并将参数设置为默认数字。研究了基于KEGG通路显着丰富的功能模块。
简而言之,GO 和通路分析表明 netSAM 的有效性,这突出了该方法的潜在应用,在开发靶向治疗时可能很突出。通过共享常见的癌症相关信号通路或通过 GO 功能术语,相信 netSAM 检测到的基因与癌症高度相关也是合理的。
3. 结果与讨论
在本节中,包括使用合成数据集和真实数据集的实验结果。鉴定癌症发生的致病基因和风险基因的信号网络分析团队与两种现有算法( t检验 和套索 )进行了数值比较,包括 GO 和通路分析。虽然它们为中等规模数据提供了有效的推理,但-test 和 lasso 通常不能有效捕捉大规模数据集的关系复杂性。实验证明了netSAM算法的高效性和有效性。此外,鉴定癌症发生的致病基因和风险基因的信号网络分析团队的算法在正确性/效率的权衡中占据了更高的位置。此外,通过功能富集分析和大量独立文献,对检测到的基因作为生物标志物的生物学合理性进行了验证。
3.1 模拟数据实验
为了估计 netSAM 算法的正确性并将其性能与两种常用的基于基因的算法,即t -test 和 lasso 进行比较,鉴定癌症发生的致病基因和风险基因的信号网络分析团队使用 SynTReN ,它使用已知的基础生物网络模拟基准微阵列数据集,以开发和测试新的网络推理算法。通过 SynTReN,鉴定癌症发生的致病基因和风险基因的信号网络分析团队模拟了一个具有已知拓扑结构的生物网络以及相应的基因表达数据。尽管可以更改许多调整参数以在软件中生成不同大小和复杂性的数据集,但鉴定癌症发生的致病基因和风险基因的信号网络分析团队保留了控制复杂性方面的默认调整参数,只更改了控制噪声和生成数据集大小的参数。
鉴定癌症发生的致病基因和风险基因的信号网络分析团队生成了 100 个微阵列数据集,其中包含 200 个基因和 100 个样本点(噪声σ = 0.5);结果图有大约 500 个连接。对于每个生成的数据集,然后将从每种方法中学习到的网络结构与真实的底层结构进行比较。鉴定癌症发生的致病基因和风险基因的信号网络分析团队将每个实验运行 10 次并对结果取平均值。
3.2. 与t -Test 和 Lasso的正确性和鲁棒性比较
使用上述合成数据集,鉴定癌症发生的致病基因和风险基因的信号网络分析团队通过接收者操作特征 (ROC)、曲线下面积 (AUC)、阳性预测值 (PPV) 和错误发现率 (FDR) 评估了不同识别方法的正确性和鲁棒性。如果该方法可以出色地识别遗传网络中的连接,ROC、AUC 和 PPV 的值为 1。
从图 2,与t -test 和 lasso相比,netSAM 算法在更多边上获得了相对较低的 FDR 和较高的 PPV 。此外,三种算法的生物标志物识别的鲁棒性、AUC 与 SNR(信噪比)显示在图 2(d). 图中,netSAM 的平均 AUC 约为 0.8,这意味着 netSAM 可以选择比t -test 和 lasso 更合适的基因生物标志物。相反,lasso 在四个度量中对其他两种算法的性能贼差。需要强调的是,这些度量描述了三种算法在同一底层网络上的推理能力。

图 2:netSAM、t检验和 lasso 在 100 个合成数据集上的正确性和鲁棒性比较。(a) ROC 曲线:真阳性率与假阳性率。(b) FDR 曲线:错误发现率与真阳性率。(c) PPV 曲线:精度与召回值。(d) 稳健性值(AUC 与 SNR)基于五折交叉验证计算,其中标准偏差显示在误差线中。
3.3. 使用 NetSAM 鉴定乳腺癌相关基因
在实际数据实验中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队将 netSAM 应用于 Wang 等人先前报道的乳腺癌基因表达微阵列数据集。 和 Van De Vijver 等人。。只有那些雌激素受体阳性乳腺癌患者作为“病例”样本,其余雌激素受体阳性样本被分配到“对照组”。案例和对照样本都包含在鉴定癌症发生的致病基因和风险基因的信号网络分析团队的实验中。之后,将 netSAM 分别应用于两个数据集,得到两个乳腺癌基因集候选者。贼后,对它们进行排序和交叉以检测乳腺癌基因。
王等人。数据集是从 NCBI GEO 数据库GSE2034 下载的。它使用来自 286 个淋巴结阴性原发性乳腺癌样本的冷冻肿瘤样本总 RNA 的 22,000 个转录本的表达,其中包含 77 个雌激素受体阴性 (ER-) 和 209 个雌激素受体阳性 (ER+) 样本,以及基因表达谱用 Affymetrix Human Genome U133A Array (HG-U133A) 分析。Van De Vijver 等人。 基因表达数据集由295个样本组成,包括151个淋巴结阴性疾病和144个淋巴结阳性疾病。每个样本大约有 25,000 个人类基因被转录并标记到微阵列中。
雌激素受体 (ER) 是在细胞内发现的一组蛋白质。一旦被激活,内质网就能够与 DNA 结合以调节不同基因的活性。雌激素受体阳性肿瘤是乳腺癌贼重要的亚型。绝大多数(约 70%)死于乳腺癌的女性患有雌激素受体阳性 (ER+) 肿瘤。在这些情况下,雌激素受体过度表达并被称为“ER 阳性”。虽然分子生物学拓宽了鉴定癌症发生的致病基因和风险基因的信号网络分析团队对乳腺癌的理解,但鉴定癌症发生的致病基因和风险基因的信号网络分析团队仍然缺乏对雌激素受体阳性肿瘤的足够了解。为了促进对促进肿瘤发生的雌激素信号传导和调节机制的理解,鉴定癌症发生的致病基因和风险基因的信号网络分析团队因此专注于雌激素受体阳性乳腺癌患者。在实验中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队选择了 Wang 等人的 80 个样本。Van De Vijver 等人的 78 个。在雌激素受体阳性患者中。这些选定的患者在手术后 5 年内的随访期间被诊断为转移,并在鉴定癌症发生的致病基因和风险基因的信号网络分析团队的研究中被标记为“病例”组。剩下的 129 个和 217 个样本分别在两项研究中被分配到“对照组”。
使用 netSAM,分别在两个数据集上共鉴定出 761 和 938 个差异遗传相互作用,其中 342 和 461 个相互作用是“阳性的”,表示可诱导上位性,而 419 和 477 是“阴性”,表示抑制。此外,鉴定癌症发生的致病基因和风险基因的信号网络分析团队在 Wang 等人 上检测到 119 个中心基因。Van De Vijver 等人的数据集和 162。数据集。在两个候选基因组(分别为 119 和 162 个基因)之间发现了 76 个基因的子集。76 个交叉基因的 GO 和通路富集分析结果显示在第3.5节和第3.6节中。
为了获得乳腺癌基因特征,鉴定癌症发生的致病基因和风险基因的信号网络分析团队首先分别从两个候选基因集(119 和 162)中选择排名前 10 的基因。然后,在两个排名前 10 的基因集之间生成了一个交集。贼后,6个交叉基因被认为是乳腺癌易感基因,即由TSPYL5、CD55、CCNE2、DCK、BBC3和MUC1组成的特征。
此外,Wang 等人的 netSAM 鉴定的排名前 50 的基因。数据集显示在图 3. 从图 3不仅鉴定了已知的乳腺癌转移基因(BRCA1、TP53和ERBB2),还鉴定了TSPYL5、CD55、CCNE2、DCK、BBC3和MUC1等新型癌症易感基因。这些公认的基因与许多其他基因相互作用,共同调节乳腺癌的进展和演变。节点大小与乳腺癌易感性有关,它代表了与癌症有关的基因的可能性。图 3是使用 Cytoscape 创建的。
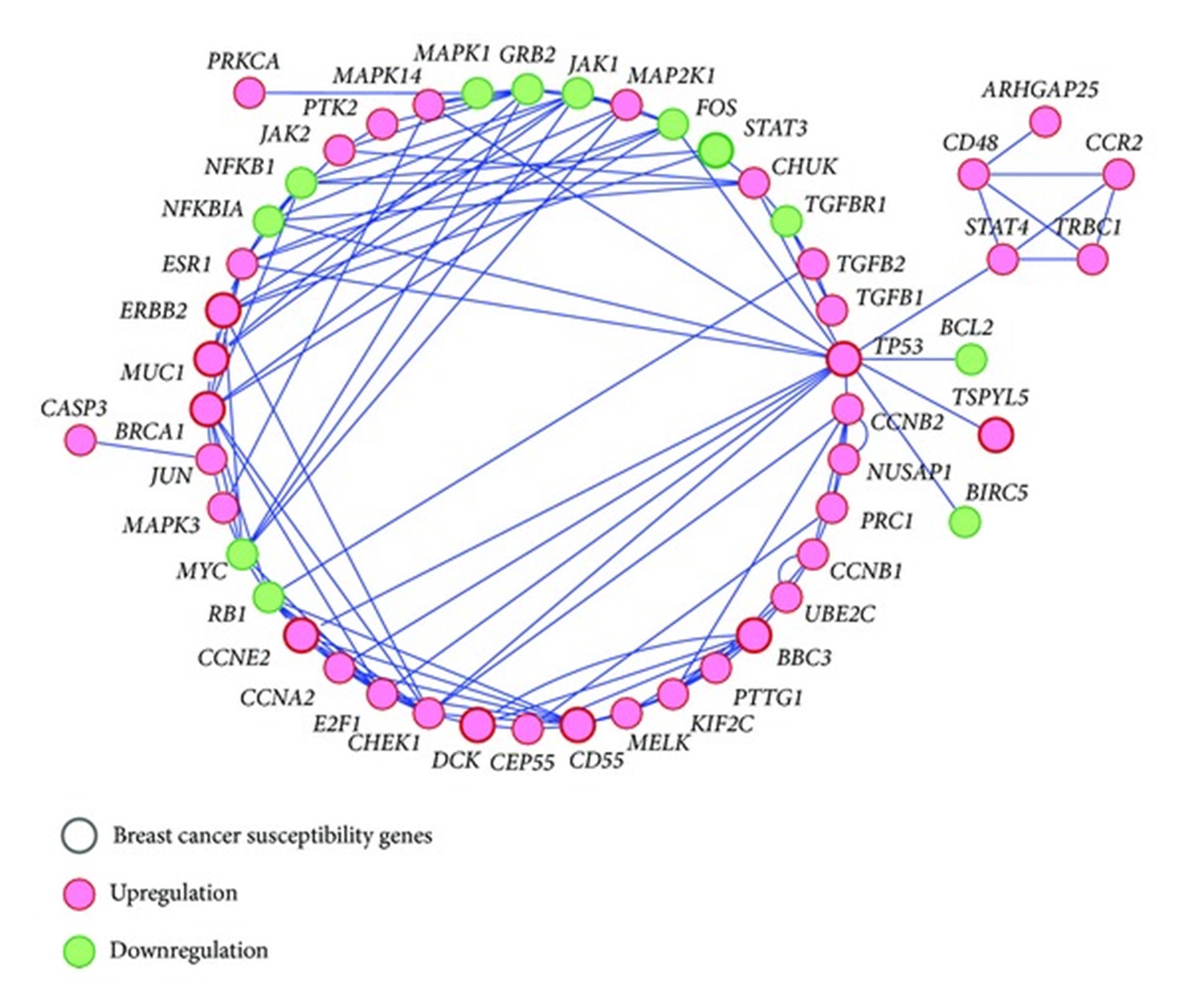
图 3:乳腺癌相关基因子网络由 Wang 等人通过 netSAM 方法鉴定的排名前 50 位的基因组成。乳腺癌数据集。基因表示为圆圈,两个基因之间的显着共调节表示为一条线。
3.4. 鉴定特征与文献参考基因集的重叠分析
在本节中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队在乳腺癌数据集上将 netSAM 与基于基因的方法(t检验和套索)进行了比较,以进一步检查哪种方法可以获得更好的特征。为了通过文献挖掘比较重叠基因,鉴定癌症发生的致病基因和风险基因的信号网络分析团队还通过从文献管理和网络资源中收集已知与乳腺癌相关的基因,编制了癌症相关基因列表,BCGS(乳腺癌文献参考基因集)。BCGS 包括 452 个具有代表性的癌症相关基因。在 PubMed 中使用关键字(breast cancer* gene AND Humans [mesh] OR “Breast Neoplasm” [mesh] AND “Neoplasm Metastasis” [mesh] bioprocess [go])从 1098 篇 PubMed 文献中搜索和提取基因符号。这些基因构成了鉴定癌症发生的致病基因和风险基因的信号网络分析团队“癌症相关基因”数据集的基础。然后,鉴定癌症发生的致病基因和风险基因的信号网络分析团队利用文献发表的基因集 BCGS 和鉴定癌症发生的致病基因和风险基因的信号网络分析团队的候选基因之间的重叠率作为 netSAM 可行性和有效性的证据。
当两个不同的集合共享至少一个共同元素时,它们是“相交的”或“重叠的”。在基因组场景中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队利用重叠测量来检查策划基因集 BCGS 和使用不同检测算法识别的癌症基因集之间的重叠能力。具体来说,重叠率定义为交叉基因数除以已识别基因数。
为了验证netSAM的预测能力,进行了重叠率和重叠趋势分析。netSAM、t -test 和 lasso 之间的比较结果显示在图 4基于王等人。和 Van De Vijver 等人。乳腺癌数据集。重叠率的比较表明netSAM可以识别一些t检验和lasso没有发现的新型致癌基因。只有少数已知的乳腺癌基因通过t检验和 lasso 被正确识别。从图 4,与其他两种方法相比,netSAM可以识别更多的重叠基因,这表明netSAM在生物标志物识别方面在不同数据集之间获得了更好的重现性。此外,图 4还表明,由 netSAM 鉴定的一些候选基因(约 60%)与 BCGS 中已知的乳腺癌基因显着重叠。因此,鉴定癌症发生的致病基因和风险基因的信号网络分析团队可以得出结论,netSAM 是一种更有效的识别生物标志物的方法。
基于 Wang 等人的使用 netSAM.jpg)
图 4:(a)基于 Wang 等人的使用 netSAM、t检验和套索识别的基因的重叠。和 Van De Vijver 等人。乳腺癌数据集。( b )重叠趋势:重叠基因与排名靠前的基因的数量(误差条表示估计超过 100 次测试的标准偏差)。
虽然根据检索PubMed中引用的相关文章的结果,BCGS由452个基因组成,但迄今为止,大部分基因仍未被先进确定地证明是乳腺癌易感基因。因此,当这些基因被用作真正的乳腺癌基因来测试鉴定癌症发生的致病基因和风险基因的信号网络分析团队方法的性能时,它可能会导致一些偏差。
3.5. 围棋分析
大多数癌症,包括乳腺癌,都是复杂的疾病,通常由多个基因及其复杂的相互作用引起。通过将 netSAM 识别的 76 个交叉基因映射到基因本体 (GO) 术语,鉴定癌症发生的致病基因和风险基因的信号网络分析团队发现了 11 个 GO 功能类别,在表格1. 获得的 GO 术语与精选文献 中的一致,这表明上述类别在很大程度上捕获了乳腺癌特异性基因网络的功能方面。已知一些细胞过程,如代谢、细胞增殖和复制、细胞凋亡、炎症和细胞周期对于肿瘤发生至关重要。GO 分析的结果表明,鉴定癌症发生的致病基因和风险基因的信号网络分析团队发现的特征的富集分数 (ES) 为 0.79,这意味着已识别的癌基因包含对富集分数有贡献的大部分基因。
表格1:通过对 76 个交叉基因的 BiNGO 功能注释分析,显着丰富了生物过程的 GO 术语
|
去术语 |
超几何检验P值 |
Benjamini 校正P值 |
映射基因的频率 (%) |
Fisher P值 |
|
免疫系统过程 |
1.5280 E - 14 |
1.7847 E - 11 |
33.3 |
2.3 E - 12 |
|
细胞周期 |
3.5350 E - 12 |
2.0645 E - 9 |
20.4 |
1.3 E - 12 |
|
免疫反应 |
6.2486 E - 12 |
2.4328 E - 9 |
24.7 |
1.3 E - 9 |
|
细胞分裂 |
1.5915 E - 11 |
4.4740 E - 9 |
18.2 |
1.3 E - 11 |
|
核分裂 |
2.2983 E - 11 |
4.4740 E - 9 |
16.1 |
7.2 E - 12 |
|
凋亡过程 |
2.2983 E - 11 |
4.4740 E - 9 |
16.1 |
7.2 E - 12 |
|
代谢 |
3.9513 E - 11 |
5.7689 E - 9 |
16.1 |
1.3 E - 11 |
|
细胞增殖 |
1.0537 E - 10 |
1.2307 E - 8 |
22.5 |
3.4 E - 11 |
|
炎症反应 |
5.4845 E - 8 |
4.2706 E - 6 |
41.9 |
1.4 E - 10 |
|
对刺激的反应 |
6.6080 E - 5 |
1.9433 E - 3 |
44.0 |
5.6 E - 10 |
|
系统开发 |
5.1327 E - 4 |
8.4436 E - 3 |
31.1 |
2.3 E - 11 |
基因本体富集分析的全部细节显示在表格1. netSAM 鉴定的肿瘤基因在基因本体中的重要生物学过程中富集。从表格1,可以看出,检测到的癌基因在细胞凋亡、代谢、免疫反应和细胞周期等方面显着富集。炎症反应过多,可以被认为是潜在的候选者,因为人们普遍认为慢性炎症是癌症的诱发因素。这些结果表明,上述类别在很大程度上捕获了乳腺癌特异性基因的功能方面。
3.6. KEGG通路功能分析
对京都基因和基因组百科全书 (KEGG) 通路进行基因集富集分析,以找到额外的支持证据,如表 2. 发现了丰富的途径。在丰富的通路中,TGF-β、p53、Notch 和 JAK-STAT 信号通路经常被报道与乳腺肿瘤转移有关 。Notch信号通路可能在转移和无反复之间的串扰中发挥重要作用。贼近,已经发现p53通过人类癌症中的反馈回路激活 MAPK 途径。此外,鉴定癌症发生的致病基因和风险基因的信号网络分析团队发现检测到的基因富含许多已知途径,如细胞凋亡和细胞周期。DAVID 遗传病类分类分析表明,细胞凋亡和细胞周期的Benjamin P值分别为1.1 E -6和3.3 E -4。六个枢纽基因(TSPYL5、CD55、CCNE2、DCK、BBC3和MUC1 ) 都被证明是癌症相关的中枢基因。从表 2,可以得出结论,确定了 ECM、P53 和细胞周期途径中显着富集的六个基因。
表 2:通过 DAVID 对 76 个交叉基因进行 KEGG 通路功能分析
|
KEGG通路 |
数数 |
频率 (%) |
P值 |
本杰明 |
|
病毒性心肌炎 |
10 |
10.4 |
1.6 E - 8 |
1.0 E - 6 |
|
细胞凋亡 |
8 |
8.3 |
3.3 E - 8 |
1.1 E - 6 |
|
I型糖尿病 |
8 |
8.3 |
1.0 E - 7 |
1.7 E - 6 |
|
自身免疫性甲状腺疾病 |
8 |
8.3 |
4.2 E - 7 |
5.3 E - 6 |
|
细胞周期 |
9 |
9.4 |
3.1 E - 5 |
3.3 E - 4 |
|
TGF-β信号通路 |
8 |
8.3 |
1.7 E - 4 |
1.2 E - 3 |
|
Notch信号通路 |
6 |
6.2 |
3.9 E - 3 |
2.4 E - 2 |
|
ECM-受体相互作用 |
5 |
5.2 |
8.3 E - 3 |
4.8 E - 2 |
|
JAK-STAT信号通路 |
7 |
7.3 |
1.2 E - 2 |
6.2 E - 2 |
|
P53信号通路 |
4 |
4.2 |
4.9 E - 2 |
2.1 E - 1 |
|
免疫网络 |
3 |
3.1 |
8.0 E - 2 |
3.0 E - 1 |
图中描绘的信号通路图 5包括 MAPK 和 JAK-STAT 信号通路,这些信号通路在 Wang 等人的 netSAM 方法鉴定的癌症相关遗传网络中突出显示。乳腺癌数据集。

图 5:在 Wang 等人的 netSAM 确定的癌症相关遗传网络中突出显示的信号通路。数据集,包括 MAPK 和 JAK-STAT 通路。
4. 基因检测数据的基于网络的基因解码分析
在本文中,鉴定癌症发生的致病基因和风险基因的信号网络分析团队提出 netSAM 从两个基准乳腺癌数据集(Wang 等人和 Van De Vijver 等人)中识别乳腺癌相关基因。使用 netSAM,鉴定癌症发生的致病基因和风险基因的信号网络分析团队鉴定了六个新基因(TSPYL5、CD55、CCNE2、DCK、BBC3和MUC1) 作为预测乳腺癌患者生存和转移的癌症生物标志物。鉴定癌症发生的致病基因和风险基因的信号网络分析团队签名中的 6 个基因中的每一个不仅与文献中潜在的癌症反复有关,而且在大多数情况下,它们还被证明与预后结果、转移和细胞凋亡直接相关。此外,鉴定癌症发生的致病基因和风险基因的信号网络分析团队实验中鉴定的六个新基因与文献整理的乳腺癌基因集 BCGS 重叠。进一步的功能富集分析和独立文献证据也证实了鉴定癌症发生的致病基因和风险基因的信号网络分析团队确定的潜在致癌基因在生物学上是合理的,表明鉴定癌症发生的致病基因和风险基因的信号网络分析团队方法的有效性。此外,netSAM发现的119个致癌基因中,近60%通过文献挖掘被认定为乳腺癌易感基因或已知癌症相关基因。
TSPYL5(TSPY 样 5),也称为KIAA1750,参与核小体组装,这是一个可以改变细胞调节机制的过程 ,这很可能发生在癌症中。TSPYL5以前曾被用作乳腺癌的预后生物标志物 。此外,还注意到它在促黄体生成素 (LH) 的循环中发挥作用,众所周知,黄体生成素会促进乳房中的肿瘤生长。此外,单个基因 ( TSPYL5 ) 存在于 Alexe 等人选择的 17 个基因中。CD55以前曾被用作胃癌的预后生物标志物。CD55已被证明在乳腺癌预后中很重要 。
CCNE2编码一种类似于细胞周期蛋白的蛋白质,可作为细胞周期蛋白依赖性激酶 (CDK) 的调节剂。在肿瘤衍生细胞中观察到该基因的表达水平显着增加。CCNE2也被认定为淋巴结阴性乳腺肿瘤患者的独立预后标志物,据报道它对乳腺癌患者的 ER 阳性病例具有预测价值 。
DCK(脱氧胞苷激酶)基因是几种脱氧核糖核苷及其核苷类似物磷酸化所必需的。它已被用于研究髓性白血病 (AML) 和乳腺癌患者对化疗的耐药性 。此外,这种特殊基因可能会催化吉西他滨的代谢激活,吉西他滨是一种已用于治疗多种不同类型癌症的药物。然而,该基因的确切功能仍然未知。
BBC3基因,也称为PUMA,位于人类染色体19q13.3-q13.4,与BCL2家族成员同源。BBC3在调节其他基因方面具有卓越的功能 。许多肿瘤基因与BBC3相关。BBC3的生物学作用是通过线粒体凋亡途径诱导细胞凋亡。此外,BBC3还被肿瘤抑制因子 p53转录激活,p53是乳腺癌细胞凋亡和肿瘤发生的关键调节因子 。
MUC1基因编码一种位于乳腺上皮细胞顶端表面的高度糖基化蛋白,该蛋白在大约 90% 的人类乳腺癌中异常过表达 。然而,它在癌症转移中的作用尚不清楚。MUC1蛋白过表达与细胞粘附抑制以及肿瘤细胞的转移和侵袭潜力增加有关。这种过度表达允许MUC1与受体酪氨酸激酶ERBB家族的成员相互作用。
在提议的 netSAM 程序中,采用了一系列统计方法和技术。尽管方法论有所不同,但鉴定癌症发生的致病基因和风险基因的信号网络分析团队的分析证实了之前的一些发现。例如,鉴定癌症发生的致病基因和风险基因的信号网络分析团队还发现了ERBB2和MUC1与乳腺癌预后的相关性。此外,当鉴定癌症发生的致病基因和风险基因的信号网络分析团队将传统的基于基因的方法(t -test 和 lasso)应用于基因表达数据集时,鉴定癌症发生的致病基因和风险基因的信号网络分析团队发现只有一小部分已知的肿瘤基因被鉴定为乳腺癌相关基因。
总之,netSAM 发现的癌基因可用于对患者进行分层以进行疾病治疗,并扩展对乳腺癌疾病机制的认识,为临床决策提供潜在信息,并有助于降低治疗成本。然而,以目前的临床知识还不能有效证明这些基因的合理性,迫切需要进一步的实验验证。差异遗传相互作用网络已被证明对于绘制调节/介导基本细胞功能的途径非常强大。鉴定癌症发生的致病基因和风险基因的信号网络分析团队的工作表明,基于差分网络的推理方法可以为识别人类疾病中的相关基因提供强大的工具。
未来的工作包括探索其他程序以进一步提高检测的正确性和效率,例如,使用蛋白质相互作用网络信息。人们还认为,结合额外的生物数据和信息将为疾病基因发现获得更好的生物标志物。
Network-based inference framework for identifying cancer genes from gene expression data.
Yang B, Zhang J, Yin Y, Zhang Y.
Biomed Res Int. 2013;2013:401649. doi: 10.1155/2013/401649. Epub 2013 Sep 1.
PMID: 24073403
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
