












【佳学基因检测】睑裂狭小综合征Blepharophimosis syndrome基因解码、基因检测
眼科疾病基因检测导读:
睑裂狭小综合征基因检测是对英文疾病名称为Blepharophimosis syndrome的眼科疾病所进行的基于基因序列变化的生理学功能而进行的检测。睑裂狭小又叫做小眼睛。该病的其他英文表达形式包括Blepharophimosis syndrome、Blepharophimosis, ptosis, and epicanthus inversus,简称为睑裂狭小综合征(BPES)。佳学基因检测通过基因解码明确了导致该病发生的基因原因,本文介绍了相关内容。
睑裂狭小综合征又叫做眼睑下垂、上睑下垂和内眦赘皮反转综合征 (睑裂狭小综合征(BPES)) 是由叉头框 L2 (FOXL2) 基因的杂合变异引起的颅面疾病。 它以常染色体显性遗传的方式沿着血缘关系进行遗传,但也可能偶尔发生。 根据突变的性质和位置。睑裂狭小综合征有两种亚型,它们都涉及相同的颅面特征:I 型,与卵巢早衰 (POF) 相关,II 型,没有全身特征。 基因型-表型相关性是佳学基因检测的重点研究内容,I 型 睑裂狭小综合征(BPES) 涉及跨越整个基因的更严重的功能变异丧失。 II 型 睑裂狭小综合征(BPES) 与导致蛋白质延长而不是功能有效丧失的移码突变有关。 已在聚丙氨酸结构域内识别出一个突变热点,该区域的确切功能需要通过基因解码进一步明确。 然而,睑裂狭小综合征(BPES) 亚型不能从遗传学上确定,鉴于相关的 POF,特别是因为患者可能仍是儿童,因此需要进行知情的遗传咨询和仔细讨论计划生育建议。 青春期后,女性患者应转诊进行卵巢储备和反应评估。 可以通过手术干预和定期监测来控制眼面部特征,以预防弱视。
睑裂狭小综合征基因检测导读:
睑裂狭小综合征又叫做上睑下垂、内眦赘皮反转综合征 (睑裂狭小综合征(BPES); OMIM #110100) 是一种罕见的常染色体显性遗传病,估计每 50,000 名新生儿中就有 1 人患病,主要影响面部中部结构的发育。 四种主要的临床症状是眼睑发育不良伴水平裂隙缩短(眼睑下垂)、上睑下垂导致垂直睑裂孔缩小(上睑下垂)、双侧皮肤褶皱从下眼睑内侧上升到上睑(内眦赘皮内翻) , 以及内侧 canthi (telecanthus) 之间的距离增加。《眼科疾病及其基因序列变化》记录了两种主要的 睑裂狭小综合征(BPES) 表型,每一种都具有四种关键的眼部症状:(i) I 型 (睑裂狭小综合征(BPES)-I),它也与 40 岁之前的卵巢早衰 (POF) 相关,包括继发性闭经,导致更年期提前和不孕症,以及 (ii) II 型 (睑裂狭小综合征(BPES)-II) 没有系统性关联。
睑裂狭小综合征(BPES) 可能由涉及叉头框 L2 ( FOXL2 ) 基因的杂合变异引起,该基因编码的转录因子主要在眼睑和卵巢的发育间充质中表达。在小鼠中,Foxl2表达定位于发育中眼睑的突出脊和卵巢滤泡细胞中。高达 75% 的受影响个体可能具有可检测到的FOXL2突变,导致单倍体不足。睑裂狭小综合征(BPES)-I 的遗传通常由男性患者向后代传递,因为受影响的女性的生育能力由于卵巢功能障碍而降低。睑裂狭小综合征(BPES)-II 可以通过男性和女性传播。
佳学基因详细研究了FOXL2基因与两种睑裂狭小综合征(BPES)类型的相关性,并报告多丙氨酸序列中最常见的变异。将描述患者在不同生命阶段的临床特征和管理,包括转诊检查卵巢储备情况。
睑裂狭小综合征致病基因鉴定基因解码:基因检测的科学依据
FOXL2基因
FOXL2是一个单外显子基因,由 2.9 kb ( NM_023067.4 ) 组成,位于染色体 3q22.3。转录的蛋白质为 376 个氨基酸,属于叉头/翼螺旋转录因子家族。FOXL2 调节许多控制细胞过程的基因,包括炎症、转录、蛋白水解、细胞凋亡和类固醇生成,包括促性腺激素。它在物种间高度保守,与人类、小鼠、大鼠、牛、山羊、猪和兔具有 100% 的同源性,并由位于 54 至 148 位的 110 个氨基酸的叉头 DNA 结合域组成。它还在第 221 位和第 234 位之间包含一个由 14 个氨基酸组成的严格保守的聚丙氨酸片段,佳学基因长期关注这一结构域对生理功能的作用。它是从 14 个丙氨酸残基扩展到 24 个丙氨酸残基的突变热点,占所有 FOXL2基因内致病性变异约 30%,主要导致睑裂狭小综合征(BPES) -II 。
FOXL2的单倍体不足是 睑裂狭小综合征(BPES) 的重要原因之一,并且是第一个涉及综合征性 POF 的常染色体基因。FOXL2可以被基因内突变以及涉及基因位点的较大基因组缺失所破坏。超过 250 个变异与 睑裂狭小综合征(BPES) 相关:FOXL2的基因内突变占 81%,可细分为插入缺失移码(44%)、框内缺失(33%)、无义(12%)、错义(11 %), 和重复。全基因缺失和包含FOXL2 的更大的亚微观缺失和邻近基因分别占分子确认病例的 12% 和 5%。
佳学基因眼科病案集截止到田02020年共收集有 460 名由于FOXL2突变而产生 睑裂狭小综合征(BPES)的情况。 最常见的变异影响两个基因内区域(图1):(i)在 12 名患者(4名睑裂狭小综合征(BPES)-II 病例和8名未定义类型)中报告了c.663_692dup p.( Ala221_Ala231dup )的聚丙氨酸区域、c .664_693dup p.(Ala222_Ala231dup) 在 5 名患者(2 名 睑裂狭小综合征(BPES)-II、2 名 睑裂狭小综合征(BPES)-I 和 1 名未定义类型)中发现,c.672_701dup p.(Ala225_Ala234dup) 在至少 80 名患者( (24名睑裂狭小综合征(BPES)-II, 2 名睑裂狭小综合征(BPES)-I,和 54名未决定疾病亚型的病例) , 和 (ii) 多脯氨酸区域, 包含第 284 到 292 位的氨基酸, 有两个重复变体: c.843_859dup p.(Pro287Argfs*75) 在 46 名患者 (3名 睑裂狭小综合征(BPES)-I, 2 睑裂狭小综合征(BPES)-II 和 41 名未定义类型) 和15 名患者 c.855_871dup p.(His291Argfs*71) (3 名 睑裂狭小综合征(BPES)-I 和 12 名未定义类型),以及 5 名c.855_871del p . ( Pro287Alafs * 71 )缺失患者(3 名 睑裂狭小综合征(BPES)-I、1 名 睑裂狭小综合征(BPES)-II 和 1 名未定义类型)。
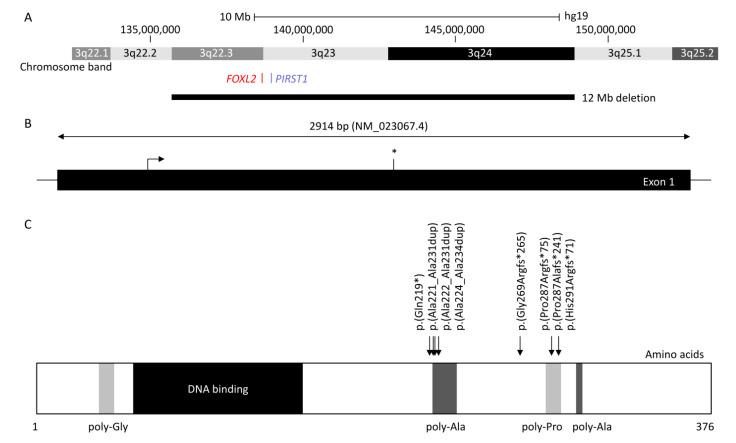
图1 展示了Forkhead Box L2(FOXL2)基因的上下游基因组位置、蛋白质结构以及多丙氨酸结构域中的热点变异。 (A) 报告了至少84例眼睑裂缝、上睑下垂和内眦上翻综合征(睑裂狭小综合征(BPES))患者中的染色体缺失,其中最大的跨越了3q22.3到3q24(12 Mb),最小的包括FOXL2或PIRST1基因(UCSC:hg19:chr3:133,064,629-153,716,375)。 (B) FOXL2由一个2.9 kb的外显子(NM_02367.4)组成,其中终止密码子用星号()表示。 (C) FOXL2是一个由376个氨基酸组成的蛋白质,由多个甘氨酸(氨基酸位置35-43)组成,用浅灰色表示,一个DNA结合蛋白或叉头结构域(氨基酸54-148)用深色表示,两个多丙氨酸(多Ala)区域(氨基酸221-234和301-304)用灰色表示,以及一个多脯氨酸区域(氨基酸284-292)用浅灰色表示(Uniprot:P58012)。在此处表示了最常见的变异(报告了五名以上患者),这些变异影响高度保守的多丙氨酸结构域(氨基酸221-234),包括c.672_701dup p.(Ala224_Ala234dup)(n = 80),c.663_692dup p.(Ala221_Ala231dup)(n = 12)和c.664_693dup p.(Ala222_Ala231)(n = 5),以及多脯氨酸结构域(氨基酸284-292),包括c.843_859dup p.(Pro287Argfs241)(n = 46),c.855_871del p.(Pro287Alafs241)(n = 5)和c.855_871dup p.(His291Argfs71)(n = 15)。其中有6例患者报告了无义变异c.655C>T p.(Gln219*)和10例患者报告了移码变异c.804dup p.(Gly269Argfs*265)。
De Baere等人之前曾提出过一种包含A-H组的近500例FOXL2内部变异的分类,旨在确定基因型与表型的相关性。然而,由于患者年龄过小或性别原因,卵巢功能通常不可用。观察到一种趋势,即导致扩展的多聚丙氨酸区域的变异,例如c.663_692dup p.(Ala221_Ala231dup)、c.664_693dup p.(Ala222_Ala231dup)和c.672_701dup p.(Ala225_Ala234dup),这些变异大多与睑裂狭小综合征(BPES)-II相关。这些变异存在家族间的可变性,其中一些病例报告与睑裂狭小综合征(BPES)-I相关。值得注意的是,一家报告了睑裂狭小综合征(BPES)-II患母亲和她的睑裂狭小综合征(BPES)-I患女儿,两者都携带c.822C>G p.(Tyr274*),揭示了可能的家族内表型变异。
FOXL2的基因内变异
FOXL2由一个外显子组成,因此,它可能像其他情况一样对无义介导的降解(NMD)具有抵抗性。因此,FOXL2中的无效变异会导致截短的蛋白质,导致叉头结构域和多丙氨酸序列的部分或有效丧失,或者由于重启翻译,产生缺少N端区域的较短蛋白质,如转染了c.157C>T p.(Gln53*)构建的COS-7细胞系。虽然在或下游的叉头结构域内的重复可以预测会产生扩展的蛋白质,但是仅涉及部分叉头结构域的突变可能会导致单等位基因不足和睑裂狭小综合征(BPES)-II,通过减少基因的转录激活活性而不影响其DNA结合。错义突变的影响可能因其位置而异,因为该基因高度保守,可能位于功能上重要的区域。大多数错义突变都映射到叉头DNA结合域,这些可能是致病的。但是,由于错义突变已在睑裂狭小综合征(BPES)-I和睑裂狭小综合征(BPES)-II中报告过,因此无法预测基因型与表型相关性。
基因解码已确认FOXL2聚丙氨酸延长序列在不同族裔家庭中存在突变热点。该高度保守区域由14个丙氨酸残基组成,二级蛋白质结构被预测为α螺旋,在突变时可能变形并破坏关键功能 。聚丙氨酸延长是已知的最常见的睑裂狭小综合征(BPES)-II突变。佳学基因病案集已经收录了8种不同的丙氨酸延长突变(c.663_692dup30 p.(Ala221_Ala231dup), c.664_693dup30 p.(Ala222_Ala231dup), c.664_701dup p.(Ala222_Ala234dup), c.667_702dup p.(Ala223_Ala234), c.672_701dup30 p.(Ala225_Ala234dup), c.684_698dup p.(Ala228_Ala232dup), c.684_698trip15 p.(Ala228_Ala232trip), c.696_728dup p.(Ala232_Ala243dup)),其中最常见的是30个碱基的重复。这些延长的重复可能是由于三核苷酸重复复制时DNA聚合酶的滑动而导致的,占所有睑裂狭小综合征(BPES)内基因突变的约33% 。虽然扩展更可能与睑裂狭小综合征(BPES)-II(无卵巢受累)相关,而截短蛋白质则与睑裂狭小综合征(BPES)-I(有卵巢受累)相关,但是一些聚丙氨酸延长,如c.664_693dup30 p.(Ala222_Ala231dup)和c.672_701dup p.(Ala224_Ala234dup),已导致一定程度的卵巢功能障碍。只有一项自体隐性同系印度家族的证据表明存在同源聚丙氨酸延长突变,c.684_698dup p.(Ala228_Ala232dup),并伴有卵巢功能衰竭;遗传带有该突变的父母和兄弟姐妹并未受到影响。因此,睑裂狭小综合征(BPES)主要被视为常染色体显性遗传疾病。
进一步进行了功能分析,以研究突变对细胞水平的影响。荧光素酶实验显示,某些无义突变(例如p.(Glu19*))会产生一个较短的蛋白质,具有一种替代起始密码子,并形成核聚集体,而野生型蛋白质在细胞核内是分散的。Caburet等人发现,突变的FOXL2多丙氨酸段重复会导致其从细胞核向细胞质的蛋白定位异常,并形成细胞质聚集体。此外,这些多丙氨酸段延长还导致与凋亡、转录调节、炎症调节、胆固醇代谢和活性氧清除等多个重要细胞过程相关的基因表达下调。基因解码提出两种解释方案,以用来说明基因突变与睑裂狭小综合征(BPES)的表型之间的关系,其中包括或不包括POF:(1)在发育的眼睑中,需要更高剂量的功能性FOXL2来靶向启动子,而在卵泡细胞中则不需要;(2)启动子中FOXL2结合位点的数量在两种组织中是相同的,但由于不同的组织特异性蛋白质组学,在眼睑中突变蛋白的聚集和定位异常比卵巢中更强。佳学基因的《眼科发育异常》病案集中还收录了一些错义突变,例如与睑裂狭小综合征(BPES)-I 或未确定类型(因为女性患者年龄较小)有关的c.931C>T p.(His311Tyr),还影响了特定基因的表达,例如类固醇原急性调节基因(STAR,OMIM 600617)。
染色体易位及FOXL2调控基因的涉及
在九名携带t(1;3)(p21;q22)易位的患者中,易位断点位于FOXL2基因及FOXL2调控基因(如PISRT1)附近。这些易位包括t(1;3)伴随3q23区域1.2 Mb的缺失、t(2;3)(q33;q23)、t(3;4)(q23;p15)、t(3;7)(q23;q32)、t(3;11)(q22.3;q14.1)、t(3;15)(q23;q25)、t(3;20)(q22;q13)和t(3;21)(q23;q22.1),与睑裂狭小综合征(BPES)相关。只有携带t(3;11)(q22.3;q14.1)的患者被认为是睑裂狭小综合征(BPES)-I 。尽管FOXL2基因本身没有突变,但是在涉及转录因子的人类遗传疾病中,例如aniridia和Axenfeld-Rieger综合征的PAX6和PITX2基因,位置效应是普遍存在的。
在FOXL2区域外的基因组区域中,例如靠近FOXL2或PISTR1的上游或下游调控区域的缺失,也被发现约占睑裂狭小综合征(BPES)的5%。涵盖3q22.3-3q24的较大缺失与未定义类型的睑裂狭小综合征(BPES)、Dandy-Walker畸形和Wisconsin综合症有关,而FOXL2或PISTR1基因的较小缺失仅导致未定义的睑裂狭小综合征(BPES)。FOXL2和PISTR1基因的上游区域在山羊、小鼠和人类中高度保守,它们的缺失导致了PIS(有角雌雄同体综合征)突变,进而导致有角山羊。该模型的特征是头颅面部缺陷、女性不育和XX性别倒错,与卵巢中FOXL2和PISTR1表达水平降低有关。荧光素酶分析表明,在FOXL2区域外的已确定的基因组缺失会影响卵巢细胞系中的基因表达。在这些区域发生的大的染色体缺失或易位等重排事件,可以使转录单元与其调控元件分离,导致与内源性突变相同的表型。在散发和家族睑裂狭小综合征(BPES)病例中发现了FOXL2上游和下游的全基因组和部分基因组缺失以及微缺失。这些缺失点分散并位于转录因子结合位点和山羊PIS基因座,需要进一步调查以充分理解调控元件。缺失在受影响的家庭成员的不同世代之间表现出减数稳定性。一些微缺失,例如FOXL2上游的197 kb缺失,已经与伴有小头畸形和智力障碍的睑裂狭小综合征(BPES)样疾病相关联。
FOXL2与原发性卵巢功能衰竭(POF)
FOXL2是哺乳动物中已知的但并非唯一的性别分化调节因子[76]。它参与胎儿发育以及成熟卵巢的维持。在产后卵巢中,FOXL2支持卵泡的生长。小鼠中FOXL2的消融导致卵母细胞发生萎缩,次级卵泡无法成熟。研究表明,标记颗粒细胞分化的STAR蛋白是FOXL2的直接靶标,作为STAR的抑制剂。结论是整个丰富丙氨酸羧基末端在FOXL2的抑制活性中很重要,并且截短变异可能会通过颗粒细胞加速分化和原始卵泡池次级耗竭而优先导致睑裂狭小综合征(BPES)和卵巢功能失调。识别出大量卵巢FOXL2靶标可能对揭示成年卵巢中FOXL2致病变异的表型效应至关重要。双基因遗传可能通过FOXL2突变和其他参与卵巢功能的基因的协同作用对睑裂狭小综合征(BPES)相关的POF做出贡献。这也可以解释FOXL2突变的表型多向性。
在睑裂狭小综合征(BPES) I型中,提示不孕的性腺激素水平的波动现象可能是部分可逆的。在睑裂狭小综合征(BPES) I型的女性患者中,不同的性腺激素水平未必一定符合POF的诊断标准。POF通常定义为40岁前出现四个月或更长时间的次级闭经和更年期后的促卵泡激素水平(FSH; >40 IU/L。然而,POF没有普遍的定义,并且由于卵巢储备的正常变异很大,因此很难诊断POF。此外,基因解码还发现,FOXL2突变的个体和仅有POF的女性在自然怀孕和促性腺激素治疗后怀孕。
临床特征
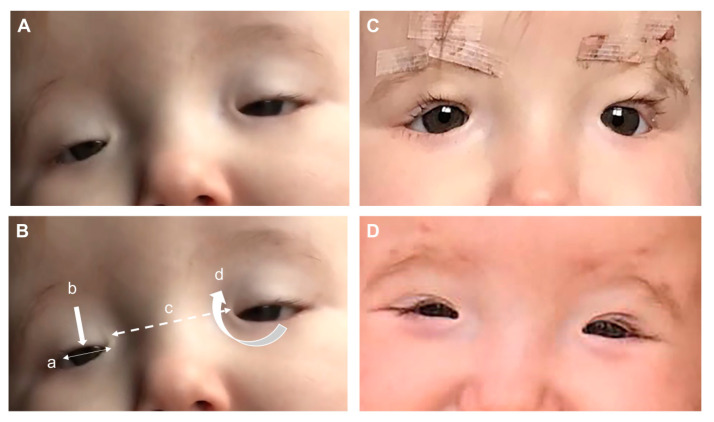
睑裂狭小综合征(BPES)主要是基于识别出四个基本特征进行的临床诊断,包括双侧眼睑发育不良,水平裂隙缩短(眼裂狭窄),上睑下垂导致上下眼裂变小(上睑下垂),由内侧下眼睑上升到上睑的双侧皮肤褶皱(内翻眼睑),以及出生时内眼角之间的距离增加(远眼角)(图2)。上睑下垂通常是双侧的,但可以不对称,提肌功能也有变异,但通常很差。眼眶骨发育正常,因此睑裂狭小综合征(BPES)患者的瞳孔间距通常是正常的。其他相关的症状并不总是出现,包括下睑翻出(外翻眼睑),泪道异常,斜视,屈光不正性弱视,宽鼻梁,厚眉弓,短人中,和向前翻转(低位)耳朵。睑裂狭小综合征(BPES)有两种表型,I型与早发性卵巢衰竭相关,而II型没有相关的全身特征。由于基因型与表型的相关性不明确,女性睑裂狭小综合征(BPES)病例应该转诊给内分泌学家或生育专家。POF的发病时间不确定,诊断也很困难,但在早期青春期时进行转诊是明智的,以尝试评估卵巢储备、卵泡计数和卵巢反应。类似外貌或早发性卵巢衰竭的家族史可以帮助诊断。
鉴别诊断
其他先天性疾病可能具有与睑裂狭小综合征(BPES)相似的特征,特别是其中两个基本特征,眼裂狭窄和上睑下垂。这些包括遗传性先天性下垂1型(OMIM#178300;常染色体显性,其中有三种亚型仅涉及上睑下垂和眼裂狭窄)、OHDO综合征(OMIM#249620常染色体显性,伴有认知障碍、先天性心脏病、上睑下垂、牙齿发育不良和眼裂狭窄;OMIM#300895 X连锁,伴有粗糙的面部特征、认知障碍和眼裂狭窄;OMIM#603736常染色体显性亚型,以前被称为Say-Barber-Biesecker-Young-Simpson综合征)、3MC综合征(OMIM#257920常染色体隐性遗传,伴有高拱眉、认知障碍、听力损失、颅缝早闭、眼球间距增大、上睑下垂和眼裂狭窄)、Noonan综合征(OMIM#163950常染色体显性,伴有身材矮小、先天性心脏缺陷、宽额头、斜下的睑裂、高拱腭、低位向后旋转的耳朵和眼球间距增大)、Marden-Walker综合征(OMIM#248700,伴有认知障碍、运动障碍、小下颌、高拱腭、腭裂、低位耳朵、驼背、关节挛缩、脑积水和眼裂狭窄)、Dubowitz综合征(OMIM#223370常染色体隐性遗传,伴有小头症、认知能力不同、上睑下垂和眼裂狭窄)和Smith-Lemli-Optitz综合征(OMIM#270400,伴有认知障碍、小头症、低张力、男性尿道下裂、多个内部器官畸形和上睑下垂)。在没有明确家族史或有任何认知障碍的情况下出现眼部面部特征时,必须考虑这些不同的鉴别诊断。虽然睑裂狭小综合征(BPES)中可能存在新生突变,但认知障碍并不是其中的特征。
治疗管理
睑裂狭小综合征(BPES)的治疗需要协调多学科团队的合作,包括儿科眼科医师和眼科整形外科医师、儿科医师、儿科内分泌学家、妇科医生、临床遗传学家和遗传咨询师。由儿科眼科医师进行视力、屈光度、斜视等方面的检查,并进行弱视的进一步治疗至关重要。眼科整形外科医师可以评估手术矫正眼部面部异常的策略,以最大程度地发挥视觉潜力。由临床遗传学家和遗传咨询师提供基因检测和咨询服务。建议将女性患者在晚童年期、青春期或早期青春期时转诊给内分泌学家或妇科医生,以评估是否出现POF,并检查卵巢储备、卵泡计数和卵巢反应等指标。
遗传咨询和基因检测
要进行全面的家族史调查和谱系分析。对于没有家族史的患者,可能是因为疾病的低渗透性、可变表达性或新生的突变。家族成员之间的疾病严重程度和预后可能不同,这种称为家族内变异性,特别是对于POF来说,这会给遗传咨询工作带来挑战,没有足够的基因解码知识可能给出错误的结果论。这种变异可能由环境、表观遗传学和/或修饰基因的影响引起。
作为常染色体显性疾病,患者的子女有50%的风险遗传到睑裂狭小综合征(BPES),但是因为睑裂狭小综合征(BPES)-I女性的不育率较高,降低了她们生育的机会,所以睑裂狭小综合征(BPES)-I更倾向于从双亲中的男性一方遗传而来。无先前家族史的患者的父母应进行FOXL2分离检测,以确定是否为新生突变,并确定有更多患儿的风险。需要考虑非生物学的解释,如其他生物学父权或未披露的领养。如果患者的兄弟姐妹没有眼睑异常,并且父母都未受影响,则患病的风险最小,尽管睑裂狭小综合征(BPES)已经被证明存在生殖细胞嵌合。
基因检测可以采用不同的方法。可以使用基于微阵列比较基因组杂交(array-CGH)的细胞遗传学检测来检测染色体异常或拷贝数变异(CNV),这对于睑裂狭小综合征(BPES)患者有很大的贡献。如果阴性,可以进行FOXL2的单基因筛查或包含该基因的定向基因组筛查。然而,全基因组测序(WGS)很可能在未来取代这种检测方法,因为它可以检测到CNV,定位于FOXL2上游或下游的基因组改变和涉及非编码调节元件的变异。
如上所述,几种综合征可能与睑裂狭小综合征(BPES)具有重叠症状,因此基因测试可以帮助澄清诊断。尽管已经有了FOXL2分子诊断,但由于缺乏高效的基因型-表型相关性,直到青春期之前仍无法区分睑裂狭小综合征(BPES)的两种类型。当儿童接受测试时,未来生殖潜力的披露是一个微妙的问题。有一个普遍的共识,即应将与纯粹有生殖影响的疾病的基因检测推迟到孩子足够大,能够理解测试的意义并自主决定是否进行测试时。然而,这个原则可能会延迟诊断澄清和及时卵巢组织冷冻,因此在决定讨论家庭计划和进行进一步的遗传咨询的年龄时必须评估这些因素。目前,应该为受影响的年轻女性提供基因咨询,包括讨论对后代的潜在风险和生殖选择。
家庭生育计划的可选方案包括自然受孕、辅助受孕、体外受精、卵巢组织采集、配子/胚胎捐赠、移植前遗传诊断、领养或选择不生育。收集原始卵泡以进行胚胎或卵子冷冻也是可能的。可以使用植前诊断或通过羊膜穿刺(妊娠15到18周)、绒毛活检(妊娠10到12周)和新型产前检测(NIPT)对胎儿细胞进行术后遗传诊断,后者使用孕妇的血液样本,其中包含胎盘带有胎儿DNA的游离DNA(cfDNA)。在遗传治疗方面取得了重大进展,CRISPR-Cas9基因编辑、基因替换或使用突变靶向药物如无意义抑制治疗等方法在未来可能适用。
早期婴儿和儿童的治疗旨在确保视觉发育完整,并预防弱视(单侧和双侧)。超过50%的睑裂狭小综合征(BPES)病例会出现一定程度的弱视,主要是由于上睑下垂和斜视引起的。三分之一的睑裂狭小综合征(BPES)病例会出现需要配戴眼镜的屈光不正。最明显的原因是由于上睑遮挡了视轴而引起的遮盖性弱视。宝宝早期通常会发展出仰头姿势以让光线进入眼睛,这并不意味着视力发育不良。因此,仅有仰头姿势并不是施行上睑下垂手术的先进指征,这种头部姿势可能会在成年时被患者忽略。与斜视相关的睑裂狭小综合征(BPES)可能还会导致双眼视觉单一发展失败,需要进行斜视手术进行矫正。通常在整个儿童视觉发育阶段(通常持续到8岁)定期进行视力评估至关重要。
如果眼睑引起遮挡性弱视,则应考虑手术方案。提升眼睑的方法有多种,使用的方法取决于儿童的年龄和提睫肌功能。在出生几周内到5岁时,在提睫肌功能较差的情况下,可以使用前额悬吊术来提升眼睑。对于轻度或中度提睫肌功能下垂症,可以使用前向或后向方法进行提睫肌修复[90]。前向方法通常被描述为超大提睫肌提升术,后向方法也涉及最大提睫肌提升。4岁以上、提睫肌功能较差的患者可以使用腱膜带(自体或异体)作为更为有效的解决方案,但如果需要处理内眼角倒睫和眼内眦距离增加,则可以将其视为分期手术的一部分。然而,与任何先天性提睫肌下垂一样,通常需要在一生中施行多次手术,应在同意过程中提及。
整形手术也可以用于矫正眼睑下垂、内眦倒置、水平睑裂大小和眼窝宽度。这些手术应该根据患者和家庭的需要进行个性化处理。如果家族中只有一个患者,那么零星的睑裂狭小综合征(BPES)病例通常在儿童早期接受整形手术。相反,具有多个家庭成员的遗传性病例可能会等待孩子长大后再做决定是否需要任何美容修复。在同意任何手术时,应该向家庭和患者解释总是有“什么都不做”的选项。在儿童早期进行矫正手术,并随后进行眼窝的持续生长,可能会比成年后进行相同手术时效果更好;然而,文献并没有描述成年睑裂狭小综合征(BPES)的重建。
两只眼睛之间的不对称也是进行美容治疗的另一个指征,旨在平衡脸部的两侧,减少不必要的关注。美容重建通常涉及两个阶段的手术,包括在3至4岁左右进行的原发性手术,纠正内眦倒睫和/或远眦距,以及在初始重建后约6至12个月进行的后续睑垂手术。在矫正内眦倒睫和远眦距方面,已应用了各种手术技术:Y-V成形术、Roveda手术、Mustardé双Z成形术和钛内眦成形术。通过进行Mustardé双Z成形术,可以通过切除皮下组织并缩短眶内侧韧带来矫正远眦距,使用缝合固定,位于眶内侧韧带插入点的后方,提供良好的美容效果,并避免使用植入物或钢丝。
早发性卵巢功能衰竭的治疗
患有睑裂狭小综合征(BPES) I型的女性患者与眼部面部畸形一起遗传不孕症。这些患者在早年可能具有正常的第二性征。如前所述,POF通常被定义为40岁之前出现四个月或更长时间的继发性闭经和绝经后的FSH水平(>40 IU/L),尽管该定义并不普遍适用。高血清FSH、黄体生成素(LH)水平升高以及雌激素和孕激素水平下降可以指示卵巢衰竭的存在。卵泡计数和对卵巢刺激的反应可能有助于评估卵巢储备。盆腔超声检查可能显示子宫发育不良,骨密度扫描可能显示骨密度降低。长期使用激素替代疗法(HRT)可缓解早期绝经症状并预防因雌激素缺乏而引起的骨质疏松症等后遗症。还应给予改善骨骼和心血管健康的建议。进一步的研究需要澄清卵巢功能是否可以从基因型预测。在有关情况不明的亲属中进行诊断也存在困难,这些亲属的睑裂狭小综合征(BPES)患病家族史和早发性卵巢功能衰竭的患病率可能是由于其他原因引起的。这强调了需要对所有睑裂狭小综合征(BPES)女性患者进行正确的卵巢功能评估的重要性,尽管在何时进行评估尚难确定。因此,应将睑裂狭小综合征(BPES)女性患者转诊至内分泌科医生和/或妇科医生进行POF评估。
睑裂狭小综合征(BPES)患者出现原发性卵巢功能不全时,可以考虑卵子刺激和提取、体外受精和医学诱导排卵。在卵巢功能不全之前储存组织是一种新的选择,涉及卵巢组织冷冻保存。如果存在正确的激素环境和正常子宫,可以将组织重新植入卵巢进行自然受孕;或者从卵巢组织中提取卵子进行体外受精,然后将胚胎植入患者的子宫或采用健康女性合法提供的基因健康卵子者的子宫。女性患者还需要个人和情感支持,以应对诊断及其对健康和关系的影响。
睑裂狭小综合征(BPES)需要一个多学科的团队,不仅在童年时期确保最大的视觉潜力,而且在成年后也需要终身监测。 FOXL2在睑裂狭小综合征(BPES)的发病机制中的作用已经得到了充分的确认,然而,遗传型与表型之间的关联仍不清楚,甚至在同一家庭谱系中也如此,这使得女性个体中POF的预测变得困难。基因咨询至关重要,新的测试方法,如全基因组测序,突出了新的突变和新的治疗方法正在被发现。从出生开始进行长期的视觉监测以预防弱视,及时治疗任何越过视轴的眼睑下垂,以及任何相关的屈光不正或斜视是防止视力丧失的关键。美容重建对于患者和家庭而言是个人的选择,但任何干预的时间都会对结果产生一定的影响,如果需要,在两个阶段的程序中可以先修复内眦上翻和眼裂宽度不一致,然后再修复眼睑下垂。青春期对于所有女性患者而言都是一个不确定的时期,何时讨论家庭计划选项尚不清楚。这应该根据每个家庭的具体情况来确定,然而,随着新技术的出现,生育能力比以往任何时候都更加强大。
- 【佳学基因检测】基因检测明确青光眼X连锁视网膜劈裂症合并闭角型青光眼:病例...
- 【佳学基因检测】一例原发性先天性青光眼合并神经纤维瘤病1型的罕见病例:病例报告...
- 【佳学基因检测】斜视基因检测该测哪些基因?斜视基因检测的价格...
- 【佳学基因检测】基因检测鉴别诊断非综合征性视网膜色素变性...
- 【佳学基因检测】视网膜母细胞瘤的基因检测及其治疗...
- 【佳学基因检测】结晶视网膜变性基因检测北京做法...
- 【佳学基因检测】眼盲基因怎么筛查?...
- 【佳学基因检测】基因治疗色盲会实现吗...
- 【佳学基因检测】单眼先天失明基因检测...
- 【佳学基因检测】眼部遗传基因检测...
- 【佳学基因检测】遗传性眼病基因检测...
- 【佳学基因检测】眼睛遗传基因哪里查...
- 【佳学基因检测】眼遗传病基因诊断方法专家共识...
- 【佳学基因检测】基因检测多少钱一次...
- 【佳学基因检测】眼睛基因检测的费用大概多少钱...
- 【佳学基因检测】基因检测价格表...
- 【佳学基因检测】结膜异常基因检测...
- 【佳学基因检测】角膜病的基因原因需要基因检测...
- 【佳学基因检测】虹膜的哪些异常会遗传...
- 【佳学基因检测】瞳孔相关的疾病基因检测...
- 【佳学基因检测】眼底疾病基因检测...
- 【佳学基因检测】基因检测脉络膜疾病...
- 【佳学基因检测】视网膜异常中的遗传性疾病基因检测...
- 【佳学基因检测】眼睑异常基因检测可以知道什么?...
- 【佳学基因检测】眼框异常基因解码机构...
- 【佳学基因检测】晶状体异常的鉴别性诊断基因检测如何做...
- 【佳学基因检测】基因检测泪腺异常有可能是哪些疾病...
- 【佳学基因检测】黄斑病状需要进行基因检测吗?...
- 【佳学基因检测】巩膜异常如何进行基因检测?...
- 【佳学基因检测】眼科基因检测的费用大概多少钱...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
