












【佳学基因检测】肾功能不全和蛋白尿经基因检测为巴林格-华莱士综合征
遗传病、罕见病基因检测导读:
巴林格-华莱士综合征基因检测是对疑似英文名称为Ballinger-Wallace syndrome的疾病进行的基于基于基因序列变化的分子诊断检测。 巴林格-华莱士综合征基因检测又叫做母系遗传性糖尿病合并耳聋。母系遗传性糖尿病和耳聋 (MIDD) 是一种线粒体遗传病,临床表现多样,可能会延误诊断。佳学基因通过线粒疾病基因检测案例介绍了一名来自陕西的患有巴林格-华莱士综合征57 岁男性帕切科(化名)患慢性肾病的病例。在患上糖尿病和耳聋之前很久,他就出现了蛋白尿;在 36 岁时,肾活检显示肾小球轻微异常,电子显微镜显示远端肾小管上皮细胞有轻度线粒体变性。二十年后,尽管没有高血压且血糖控制相对较好,但第二次肾活检显示肾硬化伴间质纤维化和小动脉透明增厚。在髓质集合管上皮中发现颗粒状肿胀的上皮细胞。电子显微镜显示在足细胞和肾小管细胞中积累线粒体,导致 巴林格-华莱士综合征的诊断。尽管没有肌肉无力,肌肉活检也显示出参差不齐的红色纤维。线粒体DNA 分析基因检测显示 m.3243A > G 突变,并开始补充牛磺酸。佳学基因致病基因鉴定基因解码的研究结果表明,线粒体功能障碍主要与进行性肾损伤有关。
基因检测案例总结性介绍
巴林格-华莱士综合征是一种线粒体遗传病,以 2 型糖尿病 (DM) 和听力障碍为特征。线粒体 (mt) DNA 的异常导致氧化能量产生缺陷引起的巴林格-华莱士综合征。巴林格-华莱士综合征的临床表现多种多样,可能会延误其诊断。mt tRNA Leu (UUR)基因中的 m.3243A > G 突变是巴林格-华莱士综合征中最常见的突变,但这种突变也见于线粒体脑肌病伴乳酸性酸中毒和中风样发作 (MELAS) 综合征。
线粒体在肾脏中非常丰富,尤其是在近端小管和肾小球上皮细胞中;线粒体功能障碍可引起肾损害。与线粒体细胞病相关的肾脏疾病包括局灶节段性肾小球硬化(FSGS) 和肾小管间质性肾病。此类并发症导致慢性肾病 (CKD) 和终末期肾病的进展,需要进行肾脏替代治疗。然而,线粒体疾病患者肾脏异质性和 CKD 进展的机制是佳学基因长期关注的焦点。
通过线粒体疾病基因解码,佳学基因检测分享一例患有巴林格-华莱士综合征的 57 岁男性帕切科(化名),他具有 m.3243 A > G 突变,并发展为慢性肾病。对于这名患者,两次肾活检相隔 20 年,以确认肾损伤的进展。肌肉活检也显示亚临床肌病。
巴林格-华莱士综合征案例介绍
一名来自陕西的 57 岁的男性因逐渐恶化的肾功能不全和蛋白尿被的主治医师转诊到佳学基因合作医院的肾脏科。他父母不是近亲结婚,属于正常妊娠生育。自 22 岁起,在他的一般健康检查中发现轻度尿蛋白从 ± 到 1 +(试纸测试)。他的第一次肾活检是在 36 岁时进行的。该标本的光学显微镜显示有 12 个肾小球,没有整体硬化;两个显示系膜基质轻度增加,其他几乎完好无损(图 1a)。 在间质中,纤维化占不到 5% 的面积,并且没有看到浸润细胞。免疫荧光未检测到 IgG、IgA、IgM、C3、C4 和 C1q。电子显微镜显示,远端肾小管上皮细胞中的一些线粒体具有失去正常结构的嵴(图 1b)。此时很难诊断病因是线粒体疾病。开始使用血管紧张素转换酶抑制剂进行治疗。
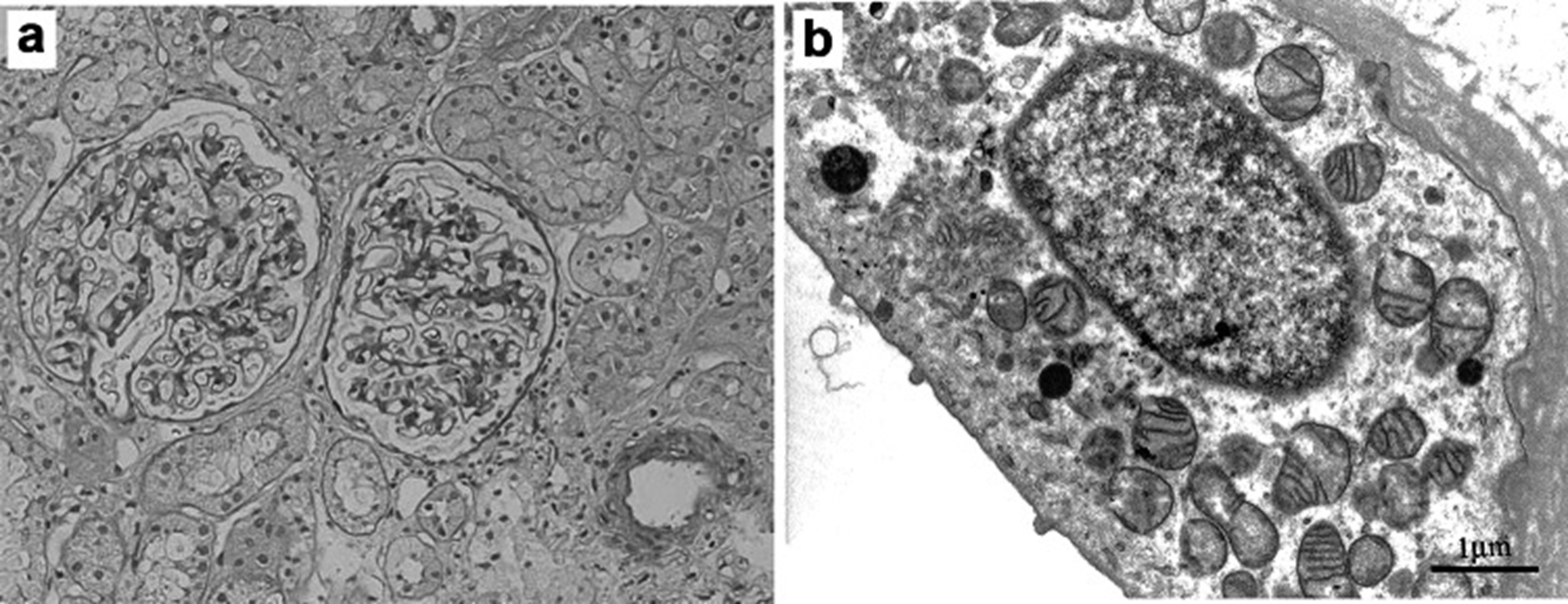
图1:最初的肾活检结果取自 36 岁。a光学显微镜显示大多数肾小球是完整的(高碘酸-希夫染色,× 200)。b电子显微镜显示远端肾小管上皮细胞中的一些线粒体具有失去正常结构的嵴
在接下来的二十年里,患者的主治医生跟踪了该患者的病情进展(图 1)。在 40 岁的体检中,他被发现患有 2 型糖尿病和双侧感音神经性听力损失。五年后,开始服药治疗2 型糖尿病,同时使用助听器。此后,血清血红蛋白 A1c (HbA1c) 水平保持在 6-8% 的范围内。尿蛋白由-增至2+,肾功能逐渐下降。他没有肌病或脑病的症状,也没有中风样发作史。他的母亲因终末期肾病(病因不明)接受了血液透析,他的弟弟患有听力损失和肾功能障碍(图3),但是,三个患病的家庭成员中没有任何一个进行过致病基因鉴定基因解码。他目前服用的药物是格列美脲、沙格列汀、二甲双胍、依那普利和普伐他汀。在 27 岁戒烟之前,他有 10 年每天吸 20 支香烟的历史。
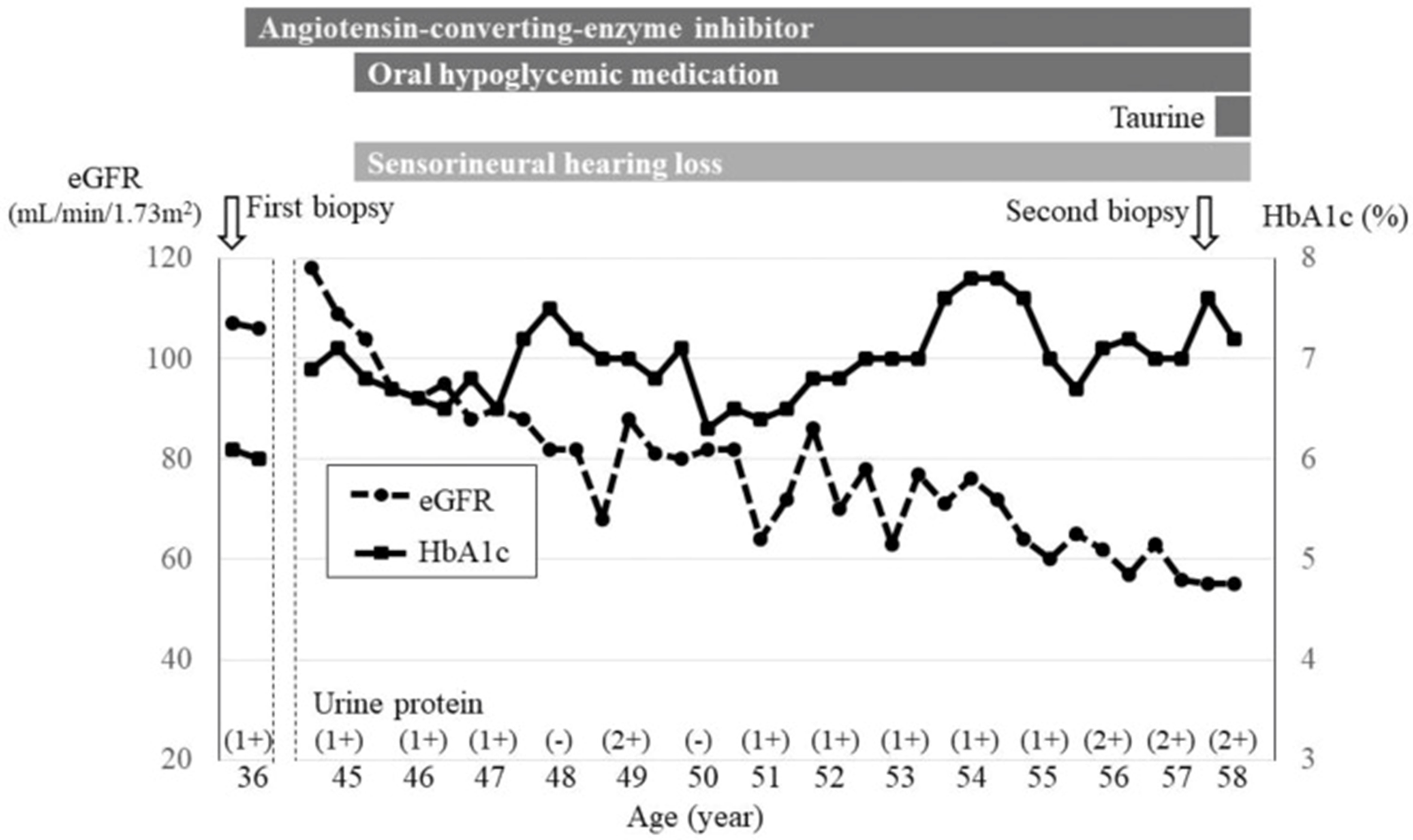
图 2:临床进程。血清 HbA1c 水平保持在 6-8% 范围内;但尿蛋白为1 + –2 + ,肾功能逐渐下降。eGFR估计肾小球滤过率,HbA1c血红蛋白 A1c
家谱.jpg)
图 3:帕切科(化名)家谱。箭头表示本报告中描述的患者。他的母亲因不明原因的终末期肾病接受血液透析,而他的弟弟则表现出听力下降和肾功能障碍
体格检查,身高160cm,体重47.8kg,BMI 18.6kg/m 2。帕切科(化名)的血压为 136/85 mmHg。帕切科(化名)的四肢没有外周水肿或肌肉无力。眼科检查未发现糖尿病性视网膜病变或高血压性视网膜病变。心电图和超声心动图均在正常范围内。从患者入院时获得的实验室数据(表1) 显示血清肌酐水平升高 (1.10 mg/dL),估计肾小球滤过率为 55 ml/min/1.73 m 2。在每克肌酐 (g/gCre) 为 0.64 g 时观察到蛋白尿;未检测到血尿(1-4 个红细胞/高倍视野)。值得注意的血清水平是 HbA1c:7.2%;乳酸:18.0 mg/dL(正常,3.7–16.3 mg/dL);丙酮酸:0.93 mg/dL(正常,0.3–0.9 mg/dL),乳酸/丙酮酸比:19(正常,< 10)。
表1:入院时血液和尿液的实验室数据
| 验血 | 钠 | 143毫当量/升 | ANA | − | |
| 白细胞 | 5300/毫升 | 钾 | 4.9毫当量/升 | PR3-ANCA | − |
| 红细胞 | 4.4 × 10 4 /毫升 | 氯化物 | 107毫当量/升 | MPO-ANCA | − |
| 血红蛋白 | 11.9 克/分升 | 钙 | 8.8 毫克/分升 | ASO | − |
| 血小板 | 17.7 × 10 4 /毫升 | 磷 | 2.6 毫克/分升 | ASK | − |
| PT-INR | 0.9 | 低密度脂蛋白胆固醇 | 79 毫克/分升 | HBs抗原 | − |
| APTT | 26.5 秒 | 血红蛋白 A1c | 7.2% | 丙肝病毒抗体 | − |
| 总蛋白 | 6.7 克/分升 | CRP | 0.78 毫克/分升 | 尿液分析 | |
| 白蛋白 | 4.2 克/分升 | 免疫球蛋白 | 780 毫克/分升 | 酸碱度 | 5.5 |
| AST | 18 单位/升 | 免疫球蛋白A | 200 毫克/分升 | 重力 | 1.024 |
| 备选方案 | 17 单位/升 | IgM | 77 毫克/分升 | 蛋白质 | 0.64 克/克Cre |
| 乳酸脱氢酶 | 158 单位/升 | 补体 3 | 122 毫克/分升 | 糖 | − |
| 碱性磷酸酶 | 353 单位/升 | 补体 4 | 32.1 毫克/分升 | 红细胞 | 1–4/HPF |
| CK | 80 单位/升 | CH50 | 48 单位/毫升 | 白细胞 | 1–4/HPF |
| 包子 | 16.8 毫克/分升 | 乳酸 | 18.0 毫克/分升 | β2-MG | 84 毫克/升 |
| 肌酐 | 1.1 毫克/分升 | 丙酮酸 | 0.93 毫克/分升 | NAG | 5.5 单位/升 |
| 尿酸 | 6.2 毫克/分升 | L/P 比例 | 19 | BJP | − |
WBC白细胞、RBC红细胞、PT-INR凝血酶原时间国际标准化比值、APTT活化部分凝血活酶时间、AST天冬氨酸转氨酶、ALT丙氨酸转氨酶、LDH乳酸脱氢酶、ALP碱性磷酸酶、CK肌酸激酶、BUN血尿素氮、LDL-cho低密度脂蛋白胆固醇、CRP C反应蛋白、Ig免疫球蛋白、CH50 50%溶血补体活性、L/P乳酸/丙酮酸、ANA抗核抗体、PR3-ANCA蛋白酶3-抗中性粒细胞胞浆抗体、MPO髓过氧化物酶、ASO抗链球菌溶血素-O、ASK抗链激酶、HBs乙肝表面、HCV丙肝病毒、Ccr肌酐清除率、HPF高倍视野、MG微球蛋白、NAG N-乙酰谷氨酸,BJP Bence Jones 蛋白
因为从帕切科(化名)的家族史怀疑有遗传性肾病,所以进行了第二次肾活检。光学显微镜显示 4 个肾小球,均显示球性肾小球硬化。在间质中,近 40% 的区域观察到慢性纤维化。还观察到含有蛋白质物质的局部慢性炎症细胞浸润和扩张的小管(图 4-a,b)。动脉和小动脉显示中度纤维内膜增厚和玻璃样变,血管平滑肌细胞增大(图 4c)。颗粒状肿胀上皮细胞 (GSEC)——据基因解码是与线粒体细胞病变相关的特定变化 ——在髓集合管上皮细胞中被发现(图4d). 与他的第一次肾活检一样,免疫荧光未检测到 IgG、IgA、IgM、C3、C4 和 C1q。电子显微镜显示中度足突消失。在一些足细胞的细胞质中观察到积累的线粒体(图 4e)和近端肾小管细胞(图 1f)。其中一部分被放大了。

图 4:57 岁时的肾活检结果。a , b光学显微镜显示整体肾小球硬化(箭头)、局部慢性炎症细胞浸润和含有蛋白质物质的扩张小管(箭头,高碘酸席夫染色,a : × 40, b : × 200 )。c血管平滑肌细胞增大(高碘酸银-乌洛托品染色,× 400)。d髓质集合管上皮中的颗粒状肿胀上皮细胞(箭头,Masson-trichrome 染色,× 400)。电子显微镜显示一些足细胞 ( e ) 和近端肾小管细胞 ( f )的细胞质中积累了线粒体,其中一些增大了(箭头)
基于这些发现,怀疑是线粒体疾病,并对他的二头肌进行了肌肉活检。改良的 Gomori 三色染色显示参差不齐的红色纤维(图 5a)。琥珀酸脱氢酶 (SDH) 染色阳性的参差不齐的红色纤维表明细胞色素 c 氧化酶染色的纤维活性不足;观察到强 SDH 反应性血管(图 5b). 这些发现与线粒体功能障碍一致。随后对肌肉标本进行的基因检测显示线粒体基因具有 m.3243A > G 突变,具有 69.9% 的异质性。因此,巴林格-华莱士综合征的诊断被认为是明确的。开始口服牛磺酸补充剂(12 克/天)。一年后,他的肾功能保持不变,没有明显的副作用。

图 5:肌肉活检的光学显微镜检查。a改良的 Gomori 三色染色上的粗糙红色纤维(箭头,× 100)。b琥珀酸脱氢酶 (SDH) 染色呈阳性的参差不齐的红色纤维(蓝色)显示细胞色素 c 氧化酶染色(棕色)的纤维活性不足(箭头,× 100)。突出显示了强 SDH 反应性血管(上图)
巴林格-华莱士综合征致病基因鉴定基因检测对蛋白尿患者诊断的启发
在该患者中,蛋白尿先于糖尿病或耳聋出现,这使得巴林格-华莱士综合征的早期诊断变得困难。尽管没有高血压或糖尿病性视网膜病变,并且血糖控制相对较好,但病理结果在 20 年内进展为肾硬化伴间质纤维化和小动脉透明增厚。虽然我们在肾小球中找不到任何 FSGS 病变,但通过电子显微镜可以在足细胞和肾小管细胞中看到聚集的线粒体。在髓质集合管上皮细胞中发现了 GSEC,这表明线粒体细胞病变。相反,未发现糖尿病肾病的典型病理表现(例如,肾小球结节或玻璃样病变)。这些观察结果表明线粒体细胞病变主要与 CKD 进展有关,
先前已在巴林格-华莱士综合征患者中报道了 CKD。尽管 DM 的病程长似乎对肾脏损害有重大影响,但巴林格-华莱士综合征患者的肾脏表现并不总是糖尿病肾病的后果。据报道,获得性线粒体功能障碍与 CKD 和动脉粥样硬化进展有关]. 线粒体细胞病变与巴林格-华莱士综合征患者 CKD 的发展相关的程度尚不清楚。然而,足细胞和肾小管细胞氧化磷酸化受损可导致活性氧过度生成以及功能和结构改变,从而导致蛋白尿、肾小管功能障碍,最终导致肾小球硬化和间质损伤。
与巴林格-华莱士综合征相关的临床特征包括早发性 DM 伴身材矮小、正常或低 BMI,以及与 DM 发病大致同时发生的听力损失 ,如本例所示。由于临床表现取决于每个器官中线粒体突变的阈值水平,因此巴林格-华莱士综合征患者会出现其他线粒体相关并发症. 我们的患者没有心脏疾病的症状或 MELAS 综合征的神经学表型,但尽管没有肌肉无力,但他的肌肉活检显示参差不齐的红色纤维。虽然线粒体疾病的临床表现因人而异,但即使没有主观症状,也可能在某些器官出现线粒体异常引起的组织学改变。
尚未确定预防巴林格-华莱士综合征并发症的治疗方法。据报道,辅酶 Q 10疗法可预防进行性听力损失并改善巴林格-华莱士综合征患者的胰岛素分泌缺陷。然而,牛磺酸修饰正常人中的第一个反密码子核苷酸 mt tRNA Leu (UUR) ,并且在来自具有 m.3243A > G 突变的 MELAS 患者的细胞的mt tRNA Leu (UUR)中没有牛磺酸修饰 . 日本最近的一项多中心、开放标签试验表明,口服牛磺酸补充剂可以有效减少中风样发作的反复,并增加MERAS 中mt tRNA Leu (UUR)的牛磺酸修饰 . 牛磺酸有望作为m.3243A > G突变的线粒体疾病引起的各种器官疾病的治疗药物。在这种情况下,除了肾素-血管紧张素系统阻断疗法外,患者还开始口服牛磺酸补充剂,以防止加重巴林格-华莱士综合征相关症状和 CKD 进展。需要仔细跟进。
总之,本病例的研究结果表明,巴林格-华莱士综合征引起的线粒体细胞病变可能与进行性 CKD 相关。如果根据临床病程怀疑与线粒体细胞病相关的肾脏疾病,主动肾活检可能会导致令人满意的诊断和治疗。
- 【佳学基因检测】线粒体疾病基因检测结果如何获得有效的治疗...
- 【佳学基因检测】 为什么线粒体病需要基因检测分子诊断?...
- 【佳学基因检测】线粒体脑肌病基因检测...
- 【佳学基因检测】胸苷激酶2缺陷症 (TK2d) 基因检测结果的多样本呈现...
- 【佳学基因检测】下颌骨发育不良早衰综合征基因检测找到发病和遗传原因...
- 【佳学基因检测】基因解码如何更好的分析线粒体基因突变检测...
- 【佳学基因检测】肥厚型心肌病与线粒体功能基因检测...
- 【佳学基因检测】线粒体疾病的基因检测与基因治疗...
- 【佳学基因检测】基因解码基因检测自噬引起的疾病,个性化治疗方案的来源...
- 【佳学基因检测】羲和号基因检测是什么水平?...
- 【佳学基因检测】过早染色体凝集率增加基因诊断如何做?...
- 【佳学基因检测】线粒体异常分子诊断怎么做?...
- 【佳学基因检测】线粒体复合物I的活性降低基因筛查怎么做才会正确?...
- 【佳学基因检测】线粒体复合物的活性降低III基因测序可以阻断遗传吗?...
- 【佳学基因检测】线粒体代谢异常如何确诊?...
- 【佳学基因检测】线粒体呼吸链活性异常如何才能不遗传?...
- 【佳学基因检测】线粒体呼吸链的活性增加风险检测在哪儿做比较好?...
- 【佳学基因检测】线粒体ATP合成酶复合物的活性降低基因分析需要怎么选择?...
- 【佳学基因检测】线粒体呼吸链的活性降低遗传风险怎样才避免?...
- 【佳学基因检测】线粒体复合物II的活性降低阻断遗传可以吗?...
- 【佳学基因检测】线粒体复合物活性降低IV基因测试怎样做才能有用?...
- 【佳学基因检测】3-甲基戊二酸尿症基因体检如何做才能正确有效?...
- 【佳学基因检测】线粒体丙酰辅酶A羧化酶缺陷风险评估采用什么样品?...
- 【佳学基因检测】丙酮酸脱氢酶复合物的活性降低遗传评估需要多少钱?...
- 【佳学基因检测】肌肉组织中线粒体异常遗传测试花钱多吗?...
- 【佳学基因检测】缺乏或没有色素b的(-245)基因测试机构怎么找?怎么联系...
- 【佳学基因检测】一胎有线粒体呼吸链缺陷,二胎会有再得吗?...
- 【佳学基因检测】3-羟酰-CoA脱氢酶水平降低是怎么遗传的?...
- 【佳学基因检测】如何才能不让线粒体赖氨酸运输缺陷遗传?...
- 【佳学基因检测】如何避免电子转移黄素蛋白-泛醌氧化还原酶缺陷遗传?...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
